
Abstract
Objectives:
The use of atypical antipsychotic medications in pediatric patients has become more prevalent in recent years. The purpose of this review is to provide a clinically relevant update of recent selected key publications regarding the use of atypical antipsychotics in this population.
Methods:
Studies reviewed included randomized, double-blind, placebo-controlled medication trials conducted within the past 5 years. A PubMed search was conducted for each of the 11 second-generation antipsychotic medications currently approved by the Food and Drug Administration for use in the United States: clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, paliperidone, asenapine, iloperidone, lurasidone, and cariprazine. Trials published in English with subjects 18 years of age and younger were included in this review. Additional studies, chosen for their significance to clinical practice, were also included at the discretion of the authors.
Results:
This review demonstrates that more empiric data are available regarding both the acute efficacy and, to a lesser extent, the longer-term efficacy and tolerability for several of the considered antipsychotic medications. The clinical conditions for which these medications have been studied include schizophrenia, bipolar disorder, Tourette's disorder, and autism spectrum disorder. They have also been used as an adjunctive treatment for disruptive behavior disorders with aggression, which have not responded to treatment with stimulants.
Conclusion:
Evidence regarding the efficacy and tolerability of antipsychotic medications for mental health disorders in children and adolescents has expanded exponentially in recent years. However, more information is needed so that evidence-based comparisons between medications can be made. In the future, data enabling the selection of medications based upon individual patient characteristics could potentially lead to greater efficacy and efficiency in treating what are frequently debilitating medical conditions. Maladaptive aggression in children, often treated with antipsychotics, is one such area in which there is a dearth of actual information available to the clinician. It is to be hoped that additional, longer-term studies of these medications will further inform evidence-based practice in clinical settings.
Introduction
A
In addition to the increasing use of antipsychotic medications in general, there is evidence that their use is increasing specifically among children and adolescents. Data obtained from the National Ambulatory Medical Care Survey (NAMCS) between 1993 and 2009 show a significant increase in overall antipsychotic use by youths, treated by both psychiatrists and nonpsychiatric physicians. During the 2005–2009 period, almost one-third of outpatient visits for a diagnosis of mood disorder (31.3%) involved the prescribing of an antipsychotic medication. Also during this period, a significant number of child and adolescent outpatient visits involving antipsychotic medication did not actually include a Food and Drug Administration (FDA) clinical indication, with a disruptive behavior disorder (DBD) diagnosis being among the most frequent diagnoses given. The most commonly prescribed antipsychotic medications during outpatient child visits were risperidone (42.1%), aripiprazole (28.0%), quetiapine (19.2%), and olanzapine (4.4%) (Olfson et al. 2012).
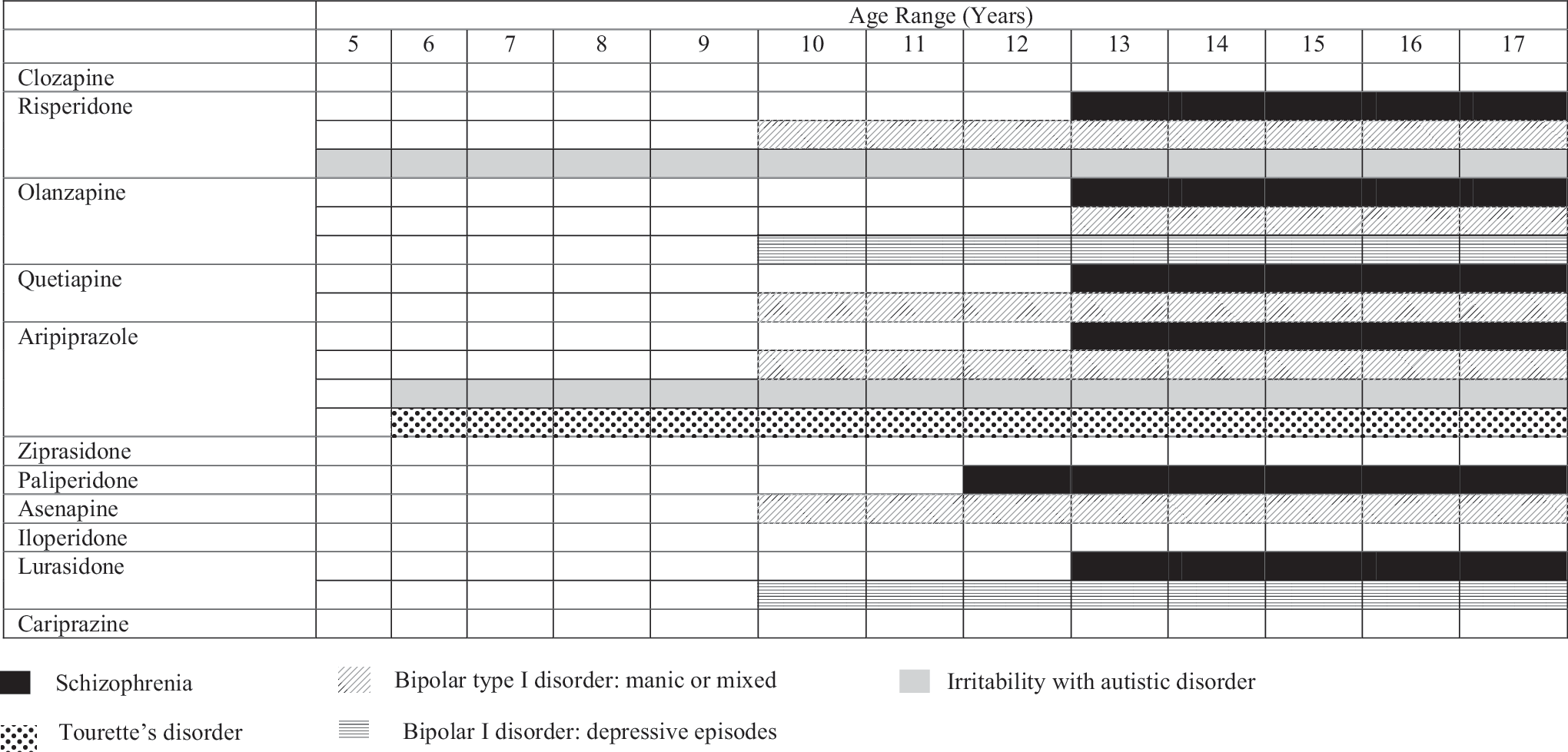
Presently, several members of the atypical antipsychotic class of medications have at least one FDA-approved indication in pediatric patients. Diagnoses for which there is an FDA approval include schizophrenia, bipolar disorder, irritability associated with autistic disorder, and Tourette's disorder (TD) (Table 1). The purpose of this review is to provide an update of recent selected key publications regarding the use of atypical antipsychotics in children and adolescents. A particular focus of this work will be to describe the clinical salience of these new data so that this information can be incorporated into clinical practice.
Methods
This review focuses on primary analyses of randomized, double-blind, placebo-controlled medication trials that have occurred within the past 5 years. A PubMed search for each of the 11 second-generation antipsychotic medications currently approved by the FDA for use in the United States was completed between November 29, 2017, and March 20, 2018, with the purpose of finding recent medication trials conducted against placebo with filtering for the following terms: clinical trial, 5 years, humans, English, and child: birth to 18 years of age. Search results were reviewed by all three authors and articles selected for their relevance to the clinical practice of child and adolescent psychiatry. Additional articles were added and reviewed at the discretion of the authors. The agents that will be considered are clozapine, risperidone, olanzapine, quetiapine, aripiprazole, ziprasidone, paliperidone, asenapine, iloperidone, lurasidone, and cariprazine.
Results
Clozapine
Clozapine is the gold standard for efficacy in treatment-resistant schizophrenia, oftentimes effective when other agents have failed, and potentially helpful in treating aggression and suicide risk in schizophrenia. It is a second-generation antipsychotic with binding affinity to serotonin 5HT2A and dopamine D2 receptors. Clozapine also binds to many other receptors (Meltzer 1994). It is not currently FDA approved for treatment in children and adolescents and we have not been able to identify any randomized or blinded studies regarding clozapine's use that have been published within the past 5 years. However, prior work has demonstrated that clozapine has a role in the treatment of children and adolescents with refractory early-onset schizophrenia. Clozapine is superior to haloperidol for both negative and positive symptoms of schizophrenia (Kumra et al. 1996) and it has also been shown to be more efficacious than olanzapine in treatment-refractory patients (Shaw et al. 2006; Kumra et al. 2008).
Clozapine presents a side effect profile with apparently more adverse effects than other drugs. Some of the side effects include nocturnal enuresis, tachycardia, hypertension, and weight gain (Shaw et al. 2006), as well as hypertriglyceridemia and a higher incidence of “pre-diabetes” (Kumra et al. 2008). Clozapine also has an increased risk for agranulocytosis, seizures, and sedation. It can cause excessive salivation and increase the risk for myocarditis (Gogtay and Rapoport 2008; Stahl 2013). Due to its risk for adverse events and the need for hematological monitoring, given its risk of agranulocytosis, clozapine's use is restricted to patients with treatment-resistant conditions.
Risperidone
Risperidone is a second-generation antipsychotic with high antagonistic affinity for 5-HT2A receptors and a moderately high affinity for D2, α-1, α-2, and H1 receptors. It was approved by the FDA for the treatment of schizophrenia in adults in 1993, and is currently FDA approved for schizophrenia in adolescents 13–17 years of age, bipolar mania in children and adolescents 10–17 years of age, and irritability associated with autistic disorder in children older than 5 years of age. Although initially synthesized in an attempt to replicate clozapine's effectiveness and lower risk profile for EPS due to its high 5-HT2A/D2 ratio, risperidone can cause EPS at higher doses. Nonetheless, its risk for EPS at low and moderate doses appears to be lower than the risk with first-generation antipsychotic drugs (Tasman et al. 2008).
Risperidone is metabolized by the enzyme cytochrome P450 2D6 (CYP2D6), which catalyzes hydroxylation of risperidone into its metabolite, 9-hydroxyrisperidone. Risperidone and 9-hydroxyrisperidone have similar pharmacological activity. The oral formulations of risperidone, which are the ones used in the studies reviewed here, are rapidly absorbed after oral administration, achieve peak plasma levels within 1 hour, and have linear pharmacokinetics. There is no food effect for risperidone, which can be administered with or without meals. See Table 2 for selected recent risperidone intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
ABC-I, Aberrant Behavior Checklist-Irritability; ADHD, attention-deficit/hyperactivity disorder; BMI, body mass index; CAARMS, Comprehensive Assessment of At-Risk Mental States; CBT, cognitive behavioral therapy; CD, conduct disorder; CGI, Clinical Global Impressions; CGI-S, Clinical Global Impressions-Severity; C-YBOCS, Children's Yale-Brown Obsessive Compulsive Scale; Hb, hemoglobin; Htc, hematocrit; MPH, methylphenidate; NCBRF D, Nisonger Child Behavior Rating Form Disruptive Behavior; ODD, oppositional defiant disorder; RBC, red blood cells; RDBPCT, randomized double-blind placebo-controlled trial; RISP, risperidone; ST, supportive therapy; TOSCA, Treatment of Severe Child Aggression; VPA, valproic acid; YMRS, Young Mania Rating Scale.
Disruptive behavior disorders
Risperidone had demonstrated efficacy in the treatment of disruptive behaviors in children of low intellect both in the short term (Aman et al. 2002) and long term (Turgay et al. 2002; Findling et al. 2004; Croonenberghs et al. 2005), as well as in children of normal intellect (Reyes et al. 2006). More recently, the use of risperidone to reduce aggression in disruptive behaviors has been explored in the Treatment of Severe Childhood Aggression Study (TOSCA), in which the children carried a diagnosis of a DBD (Aman et al. 2014). This study consisted of a 3-week open-label trial of methylphenidate combined with a behavioral intervention that enrolled children 6–12 years of age with severe aggression, including physical harm. For those who did not respond to these two treatments in combination, participants were then eligible to enter a 6-week double-blind placebo-controlled study in which patients were randomized to receive adjunctive treatment with either risperidone or placebo. The mean final dose of risperidone was 1.7 ± 0.75 mg/day.
When compared to the placebo group, risperidone provided moderate, but variable improvement in aggression and other disruptive behaviors when added to the basic treatment with parent training and stimulant medication, as measured by the Nisonger Child Behavior Rating Form (NCBRF) Disruptive–Total subscale, the NCBRF Social Competence subscale, and Antisocial Behavior Scale Reactive Aggression subscale. Adverse events included prolactin level elevations and gastrointestinal upset, which occurred more frequently in the group augmented with risperidone. Weight gain in this group was relatively modest.
Following the initial study, Findling et al. (2017a) conducted a study looking at clinical responders of the 9-week TOSCA trial, who had presented with improvement in the Clinical Global Impressions-Improvement (CGI-I) scale plus substantial reduction in parent ratings of disruptiveness. This study observed the patients for another 12 weeks (21 weeks in total), while they remained on blinded treatment. The main purpose was to check the durability of the acute phase positive responses at week 21 and the accumulation of side effects. The findings indicated that those children who responded at week 9 continued to respond over the following 12 weeks regardless of what treatment arm they were originally assigned. The 103 extension participants showed no significant between-treatment group outcome differences. This may have been because participants in the extension were selected for good response.
Finally, Gadow et al. (2016) conducted a prospective follow-up study of the TOSCA trial 12 months after the initial baseline evaluation. This study compared placebo group and the group augmented with risperidone. Both randomized groups improved from baseline to follow-up, but the parent-reported behavioral outcomes showed no significant differences between groups. Nonetheless, exploratory findings for secondary outcomes suggested that there was a greater benefit from augmentation with risperidone. Prolactin levels were significantly higher in the group treated with risperidone.
Secondary analyses of the TOSCA study have indicated that risperidone might be useful in reducing context-specific severity of attention-deficit/hyperactivity disorder (ADHD) and oppositional defiant disorder (ODD) symptoms, peer aggression, and symptom-induced impairment (Gadow et al. 2014). In addition, greater baseline severity has been associated with better treatment response, either with stimulant and parent training or in their combination with risperidone in a subsequent study on predictors and moderators (Farmer et al. 2015).
Arnold et al. (2015) examined the effect of augmented treatment on the severity of anxiety, mood, autism spectrum disorder (ASD), and schizophrenia spectrum disorder (SSD) symptoms, and found that the addition of risperidone improved symptoms and functioning in these areas. The SSD symptoms did not include hallucinations or delusions, but social withdrawal and disorganized impulsiveness. Therefore, the improved symptoms were considered to fall into the group of anxiety-social avoidance rather than the psychotic symptom spectrum. Anxiety also mediated improvement in DBD, suggesting an anxiety-driven fight or flight response in DBD with aggression.
A study conducted by Jahangard et al. (2017), using a similar design to the TOSCA trial, studied subjects with ADHD and ODD diagnoses who failed to have adequate clinical improvement after a standard treatment with methylphenidate (15–20 mg/day) and family therapy. The study was an 8-week double-blind, placebo-controlled randomized trial in which risperidone at doses of 0.5 mg/day was used as an adjuvant treatment to methylphenidate in children 7–10 years of age. Symptoms of ADHD rated by both parents and experts decreased over time, but more so in the group augmented with risperidone. However, the effect sizes for symptom improvement with risperidone were small to medium. Adverse effects included weight gain and higher prolactin levels. Overall, side effects increased over time in the group augmented with risperidone.
Autism spectrum disorder
The efficacy of risperidone for the treatment of ASD symptoms such as irritability and aggression has been demonstrated in the past (McCracken et al. 2002; Shea et al. 2004; Sharma and Shaw 2012). More recently, Kent et al. (2013) examined the efficacy and safety of two different risperidone doses in children and adolescents 5–17 years of age, with a diagnosis of ASD. This 6-week double-blind, placebo-controlled multicenter study randomized participants to one of two fixed weight-based doses of risperidone or placebo: risperidone low dose was 0.125 or 0.175 mg/day depending on the subject's weight (< or ≥45 kg) and high dose was 1.25 mg/day (20 to <45 kg) or 1.75 mg/day (≥45 kg).
Irritability scores as measured by the Aberrant Behavior Checklist-Irritability (ABC-I) as well as global functioning, obsessive compulsive symptoms, and hyperactivity improved in the high-dose risperidone group and separation from placebo was observed from day 8. The low-dose group showed improvements in stereotypic behavior when compared to placebo. A higher percentage of patients in the placebo group were treated with antihistaminic drugs. Adverse effects included somnolence, sedation, increased appetite, and increased weight, and were more frequent in the high-dose versus the low-dose or placebo groups. Extrapyramidal symptoms (akathisia) and changes in mean prolactin and insulin levels were greater in the high-dose group.
Other updates include an uncontrolled follow-up reassessment 21.4 months (1.8 years) after initial entry into the 8-week placebo-controlled randomized trial conducted by the Research Units on Pediatric Psychopharmacology (RUPP) Autism Network (McCracken et al. 2002) to assess the safety and tolerability of risperidone in children and adolescents 5–17 years of age with autism and severe irritability (Aman et al. 2015). The mean risperidone dose used was 2.47 mg/day (SD = 1.29 mg). The study found that the risperidone group presented with improvement in social skills and other maladaptive behaviors such as aggression, self-injury, hyperactivity, and agitation, as well as a decrease in irritability and sensory problems. Noteworthy adverse effects included enuresis, increased appetite, and weight gain.
Another study, based on data collected through the RUPP multisite study, explored the role of baseline symptom severity in the efficacy of risperidone (Levine et al. 2016). Symptom reduction increased with moderate through severe baseline severity. Those with moderate to severe baseline ASD symptoms demonstrated decreased irritability and lethargy on the ABC parent ratings with risperidone. There were no changes in clinician ratings of irritability or CGI, nor were there any changes on parent ratings of stereotypic behaviors, hyperactivity, or inappropriate speech.
Bipolar disorder
In the area of mood disorders, previous research showed risperidone monotherapy to be effective in the acute treatment of mania in children and adolescents with bipolar disorder (Haas et al. 2009a). More recently, Kowatch et al. (2015) conducted a study with younger children 3–7 years of age with a diagnosis of bipolar I (BP-I) disorder during a mixed or manic episode, psychotic or nonpsychotic, and a score of ≥20 on the Young Mania Rating Scale (YMRS) at the time of randomization. The study had three arms comparing risperidone, valproic acid (VPA), and placebo. The mean dose of risperidone at endpoint was 0.5 mg/day (range of 0.5–0.75 mg/day) and the mean dose of VPA was 300 mg/day. YMRS total scores improved in risperidone-treated subjects versus placebo, but not in VPA versus placebo-treated subjects. Adverse effects for those treated with risperidone included increased prolactin levels and elevated unconjugated bilirubin, GGT, and body mass index (BMI). There were also decreases in albumin and total protein, and elevations in cholesterol and insulin. In the group treated with VPA, there was an increase in cholesterol and weight/BMI, a decrease in albumin, total RBC, hemoglobin, and hematocrit, and an increase in mood lability.
Schizophrenia/psychosis
Risperidone is efficacious in the treatment of symptoms of schizophrenia in adolescents (Haas et al. 2009b, 2009c). More recently, to study its potential role as a preventive intervention for psychosis, McGorry et al. (2013) studied subjects with a clinical phenotype at ultra-high risk for psychosis, between 14 and 30 years of age. This 12-month double-blind, placebo-controlled randomized trial presented three alternatives: low-dose risperidone and cognitive behavioral therapy (CBT), CBT and placebo, and supportive therapy and placebo. Risperidone was started at 0.5 mg/day and increased over 4 weeks up to 2 mg/day, if tolerated. There were no statistically significant differences between the three groups in the rates of transition to full-threshold psychosis over 12 months. All groups improved, particularly in terms of negative symptoms and overall functioning. These results failed to provide support for the first-line use of antipsychotic medications in patients at ultra-high risk of psychosis, suggesting that an initial approach with supportive therapy is likely to be effective with fewer risks.
Olanzapine
Olanzapine is currently FDA approved in children and adolescents for use in the treatment of schizophrenia (Kryzhanovskaya 2009), manic or mixed episodes as both monotherapy and in combination with lithium or valproate (Tohen et al. 2007), and bipolar depression (Detke et al. 2015). However, due to its risk for weight gain, (Olanzapine Package Insert 2009) it is recommended that prescribers carefully consider olanzapine's risks, while also considering other medications for the treatment of their teenage patients. The pharmacokinetic profile of olanzapine has been found to be similar to that described in adults, with no adjustments needed in dosing (Lobo et al. 2010). A study by Theisen et al. (2006) found that, consistent with other studies, olanzapine concentrations were lower in smokers than in nonsmokers, suggesting a higher rate of clearance. See Table 3 for selected recent primary olanzapine intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
BP-I, bipolar I disorder; CDRS-R, Children's Depression Rating Scale-Revised; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; YMRS, Young Mania Rating Scale.
Bipolar depression
A study by Detke et al. (2015) investigated the effect of olanzapine/fluoxetine combination (OFC) treatment for BP-I depression and found that, after 8 weeks of randomization to either OFC (flexible dosing, target dose 12/50 mg) or placebo, OFC was statistically superior to placebo, with more patients achieving response (78.2% vs. 59.2%) at a faster rate (median of ∼3 weeks vs. ∼5 weeks), as well as remission (59.0% vs. 43.4%). The OFC group also demonstrated greater improvement on all secondary measures of depressive symptomatology and overall illness severity. The authors concluded that for this study, OFC was superior to placebo in the acute treatment of BP-I depressive episodes. However, it should be noted that the weight gain seen with OFC was 4.4 kg, whereas the weight gain observed with placebo was 0.5 kg during the course of study participation.
Posthoc analyses
Of the three posthoc analyses that are included herein, two studies looked into whether early treatment response was predictive of treatment outcome in youth being treated with olanzapine.
The first study by Stentebjerg-Olesen et al. (2015) found that early response (ER) at week 3 had slightly better predictive power than ER at week 2 for both ultimate response (UR) and remission in adolescents with schizophrenia, although both were significant, and a threshold for ER of ≥20% reduction on the Brief Psychiatric Rating Scale for children (BPRS-C) had the best predictive validity for UR. It was also noted that most patients who were ultimate nonresponders failed to demonstrate a response at week 2 or 3.
The second analysis by Xiao et al. (2017) found that significantly more patients with BP-I disorder who achieved ER status by week 1 went on to achieve UR at week 3 and remission at study endpoint. Receiver operating characteristic (ROC) curves determined that a cutoff threshold of 35.5% reduction in YMRS total score at week 1 showed the highest accuracy (70.2%) and the greatest area under the curve (0.75) in predicting UR.
Finally, the third posthoc analysis by Kemp et al. (2013) examined the associations between obesity, acute weight gain, and response to treatment with olanzapine in adolescent schizophrenia. In addition to significantly more olanzapine-treated patients gaining ≥7% of their body weight at any time (45.8% vs. 14.7%), the BPRS-C total scores demonstrated significant improvement for olanzapine-treated patients compared to placebo and were decreased by a greater percentage in those with significant weight gain compared to those without in both the olanzapine group (45.6% vs. 31.9%) and the placebo group (34.1% vs. 16.8%). Despite this significant inverse correlation, the authors found that the relationship became nonsignificant when the analyses were controlled for treatment duration, which was itself significantly related to increased weight and improvement in symptoms. It was suggested that patients who experience improvement with olanzapine may be more likely to continue treatment and experience weight gain secondarily.
Quetiapine
Quetiapine is FDA approved for use in children/adolescents with schizophrenia (Findling et al. 2012a) and acute mania (Pathak et al. 2013) as both monotherapy and adjunct to lithium or divalproex. The binding profile for quetiapine differs slightly from other atypical antipsychotics, in that, it has weaker antagonistic effects at dopamine D2 and serotonin 5-HT2 receptors, stronger antagonistic effects at α-1 receptors, and modest histaminergic effects (Nasrallah 2008). The pharmacokinetic profile has been described as being similar to that of adults (McConville et al. 2000) with no dosage adjustments required for pediatric patients (Winter et al. 2008). See Table 4 for selected recent primary quetiapine intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
BP, blood pressure; CDRS-R, Children's Depression Rating Scale-Revised; CGI-BP, Clinical Global Impressions-Bipolar; CGI-I, Clinical Global Impressions-Improvement; PANSS, Positive and Negative Syndrome Scale; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; YMRS, Young Mania Rating Scale.
Bipolar mania
As a treatment for children and adolescents with BP-I mania, Pathak et al. (2013) found in their 3-week trial that quetiapine (dosed at 400 or 600 mg/day) demonstrated significantly greater improvement over placebo in manic symptoms and equal efficacy in patients subcategorized by age (10–12 years vs. 13–17 years), gender, ADHD status, and psychostimulant use. Mean change in body weight was greater in the quetiapine groups than with placebo, but there were few incidences of potentially clinically significant shifts in laboratory, hematologic, and vital sign measures. It was concluded that both quetiapine doses were significantly more effective for acute manic symptoms in youth with BP-I disorder, and that the safety profile in children was consistent with that of adults with bipolar disorder.
Bipolar mania/schizophrenia
Many of the individuals from the previous study went on to participate in a continuation study by Findling et al. (2013d) looking into the safety, tolerability, and efficacy of quetiapine in youth with both schizophrenia and BP-I disorder. The 26-week treatment course was completed by 237 (62.2%) of the enrolled patients (71.0% schizophrenia and 54.6% bipolar disorder) with the mean dose of quetiapine and duration of time being 632 mg and 156 days in the schizophrenia subgroup, and 571 mg and 137 days in the bipolar disorder subgroup. The most common adverse events, similar to the adult profile of bipolar disorder patients, included somnolence, headache, sedation, and vomiting. Weight increase (mean change in BMI of 0.9 kg/m2) and increased appetite were also seen in this pediatric population, and it was observed that a number of adverse events were more frequently seen in patients from the prior-placebo rather than the prior-quetiapine group in the previous trials, possibly indicating improved tolerability over time.
While patients treated with quetiapine during the acute studies continued to demonstrate significant improvement in symptoms and functioning, improvement was generally greater in prior-placebo patients. It was concluded that quetiapine at the aforementioned dosage and duration is safe and generally well tolerated in these pediatric populations.
Bipolar depression
Findling et al. (2014b) also studied the efficacy and safety of extended-release quetiapine fumarate for bipolar depression in children 10–17 years of age. When flexibly dosed between 150 and 300 mg/day, improvement was seen in both quetiapine XR and placebo groups, with subgroup analyses showing consistency for patients with or without rapid cycling, with BP-I or BP-II disorder, and in children 10–12 or 13–17 years of age. Rates of response and remission from baseline were 63.0% and 45.7% for quetiapine XR and 55.0% and 34.0% for placebo. It was concluded that quetiapine XR did not demonstrate efficacy relative to placebo in this study, but that the medication in this dose range was generally safe and well tolerated.
Aripiprazole
Aripiprazole is currently FDA approved in children and/or adolescents as a treatment for schizophrenia (Findling et al. 2008a), acute treatment of manic and mixed episodes associated with BP-I disorder (Findling et al. 2009), irritability associated with autistic disorder (Marcus 2009; Owen et al. 2009), and TD. It has a strong affinity for the dopamine D2 and D3 receptors and moderate affinity for D4 receptors, with partial agonism occurring with the D2 receptors (Whitney et al. 2015). At similar doses, the maximum plasma concentrations at steady state were found to be higher in the pediatric population compared to adults and also required less time to achieve (Mallikaarjun et al. 2004). The safety profile has been found to be similar to that of adults (Findling et al. 2008b). See Table 5 for selected recent primary aripiprazole intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
BP-NOS, bipolar disorder not otherwise specified; CGI-S, Clinical Global Impressions-Severity; CGI-TS, Clinical Global Impressions-Tourette Syndrome; NNT, number needed to treat; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; YGTSS-TTS, Yale Global Tic Severity Scale–Total Tic Score; YMRS, Young Mania Rating Scale.
Tourette's disorder
There were two studies investigating aripiprazole as a treatment for TD. The first by Yoo et al. (2013), studying the effect of aripiprazole dosed up to 20 mg/day in children 6–18 years of age, demonstrated a significant reduction in tics compared to placebo. Mean decreases in the phonic tic score and the Tourette's Syndrome CGI-Severity (CGI-S) of Illness score were larger in the active medication group compared to placebo, with no difference being found on the motor tic score between both groups. The active treatment group had an endpoint response rate of 65.6% versus 44.8% with placebo. The data suggest that fairly rapid symptom improvement might be achieved at relatively low doses, as nearly 80% of the total efficacy response was achieved within 6 weeks at doses of 10 mg/day or less.
The second study by Sallee et al. (2017) investigated the effects of both low-dose (<50 kg at baseline, 5 mg/day; ≥50 kg at baseline, 10 mg/day) and high-dose (<50 kg at baseline, 10 mg/day; ≥50 kg at baseline, 20 mg/day) aripiprazole in children 7–17 years of age and demonstrated statistically significant improvement in tics for both low-dose and high-dose aripiprazole groups (45.9% and 54.2% decrease from baseline scores, respectively) compared to placebo. The magnitude of improvement for most efficacy endpoints was generally greater with high-dose versus low-dose aripiprazole. The medication was frequently well tolerated and the study concluded that aripiprazole might provide an effective and safe treatment option for those with tolerability concerns.
Autism spectrum disorder
Findling et al. (2014a) sought to expand on previous medication trials of aripiprazole as a treatment for irritability associated with autistic disorder by studying its safety and efficacy for maintenance purposes in 157 children between 6 and 17 years of age. After a stabilization phase (13–26 weeks of single-blind aripiprazole treatment, flexibly dosed between 2 and 15 mg/day), those participants demonstrating a 12-week period of stable response to medication then entered a randomization phase (up to 16 weeks of double-blind treatment with continued aripiprazole or placebo). The mean time to relapse for 25% of the group occurred at 56 and 29 days for aripiprazole and placebo respectively, which was not found to be statistically significant. Of note, a posthoc analysis found a clinically relevant number needed to treat six to prevent one additional relapse.
Bipolar disorder
A study that examined the postacute efficacy of aripiprazole in the treatment of pediatric bipolar disorder examined participants who continued blinded treatment after completing an acute 4-week, double-blind placebo-controlled study (Findling et al. 2009, 2012b). The authors found that for patients between 10 and 17 years of age, treated with 10 or 30 mg/day for up to 30 weeks, aripiprazole demonstrated statistically significant greater improvement in manic symptoms when compared to placebo over this period of time. The aripiprazole 10 and 30 mg/day treatment groups had a significantly longer median time to discontinuation from the point of randomization (15.6 and 9.5 weeks, respectively, vs. 5.3 weeks with placebo), as well as being significantly superior in terms of response rates (58.7% and 64.8% vs. 29.7% in placebo) and Children's Global Assessment Scale (CGAS) and CGI-bipolar severity of overall and mania scores than those who received placebo.
A second study (Findling et al. 2017b) investigated the use of aripiprazole in treating symptomatic youth at familial high risk for bipolar disorder (parent diagnosis of bipolar disorder and another first- or second-degree relative with a mood disorder), who had failed to respond to psychotherapy. These symptomatic, at-risk patients had been previously described as having “cyclotaxia.” Participants 5–17 years of age were treated with up to 15 mg/day of aripiprazole (mean total daily dose 7.1 mg/day) and demonstrated significantly greater improvement in manic symptoms, overall bipolar symptom severity, and functioning scores compared to placebo. The study found that aripiprazole was generally well tolerated and demonstrated significant efficacy in reducing subsyndromal symptoms of bipolar disorder in children and adolescents with cyclotaxia.
Schizophrenia
There was one study addressing aripiprazole as a maintenance treatment in adolescent schizophrenia by Correll et al. (2017). After being stabilized on 10–30 mg/day of aripiprazole, participants between 13 and 17 years of age were randomized to receive medication or placebo for up to 52 weeks. Aripiprazole treatment was associated with a significantly longer time to (hazard ratio = 0.46) and fewer participants (19.4% vs. 37.5% with placebo) meeting criteria for exacerbation of symptoms/impending relapse. Time to discontinuation was significantly longer and tolerability in the active treatment group was consistent with the known profile of aripiprazole. It was concluded that the medication was a safe and effective maintenance treatment option for this population.
Posthoc analyses
One posthoc analysis article studied the relationship between prior antipsychotic exposure (PAE), weight change, and adverse events in pediatric patients receiving aripiprazole for the treatment of irritability in autism (Mankoski et al. 2013). That study found that compared with those having had PAE, antipsychotic-naive (AN) patients had greater mean changes in weight compared to placebo (treatment difference of 1.2 kg with AN vs. 0.9 kg with PAE). The trend suggested that younger subjects with higher baseline weight z-scores were at greatest risk of weight gain and AN patients were at higher risk of a new-onset event related to somnolence.
Two of the studies addressed how to better assess treatment response with aripiprazole in the treatment of pediatric BP-I disorder. Findling et al. (2013c) found that the effect sizes on the 10-item parent General Behavior Inventory Mania (GBI-M10) total score were similar to those of the clinician-rated scales, indicating the possible value of this parent-completed scale in detecting symptom change in both research and clinical settings. The same data set was also used by Youngstrom et al. (2013) to compare multiple outcomes measures using a range of definitions of response to evaluate which might be most clinically relevant. The authors concluded that clinically meaningful definitions included ≥50% reduction in YMRS total score and a composite measure of response (YMRS <12.5, Children's Depression Rating Scale-Revised (CDRS-R) ≤40, and CGAS ≥51), with parent-reported measures being better indicators of symptom change than those based on subject self-report.
Finally, Correll et al. (2013) investigated whether early response (ER) or nonresponse (ENR) at week 2 or 3 of treatment with aripiprazole in adolescents with schizophrenia predicts clinical outcome. This analysis found that ER/ENR status at week 3 provided the best overall sensitivity, specificity, and accuracy in predicting a decrease of at least 30% in Positive and Negative Syndrome Scale (PANSS) total score by week 6. The authors noted that if a different dosing/titration schedule was used, this could change the predictive time point in actual clinical practice.
Ziprasidone
Ziprasidone is a second-generation antipsychotic approved for adults in the treatment of schizophrenia and BP-I disorder, but with no current FDA-approved indications for children and adolescents. Ziprasidone is a serotonin 5-HT2A and dopamine D2 antagonist with a higher 5-HT2A/D2 receptor affinity in vitro than other atypical antipsychotic agents. It exhibits potent interactions with 5-HT2C, 5-HT1D, and 5-HT1A receptors. Ziprasidone has low affinity for α-1adrenoceptors, histamine H1, and muscarinic M1 receptors (Stahl and Shayegan 2003). Its oral form reaches peak plasma concentrations at 6–8 hours and its absorption increases with food up to twofold (Miceli et al. 2007). As such, ziprasidone should be taken with food. The major CYP contributor to the oxidative metabolism of ziprasidone is CYP3A4. Ziprasidone has a higher risk and a label warning for QTc prolongation (Orsolini et al. 2016) and Torsade de pointes (Kelly and Love 2001). Prior research has reported ziprasidone to be effective for the treatment of tics in adolescents with Tourette's syndrome (Sallee et al. 2000). See Table 6 for selected primary ziprasidone intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
ADHD, attention-deficit/hyperactivity disorder; BP-I, bipolar I disorder; BPRS-A, Brief Psychiatric Rating Scale-Anchored; CGAS, Children's Global Assessment Scale; CGI-I, Clinical Global Impressions-Improvement; CGI-S, Clinical Global Impressions-Severity; OLE, open label extension; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; YMRS, Young Mania Rating Scale.
Schizophrenia
Findling et al. (2013b) evaluated the short- and long-term efficacy, safety, and tolerability of ziprasidone in adolescents 12–17 years of age with schizophrenia in a 6-week international, double-blind, multicenter, randomized placebo-controlled trial followed by a 26-week open-label extension (OLE). Subjects were randomized in a 2:1 ratio to flexible-dose oral ziprasidone (40–160 mg/day, based on weight) or placebo. The starting dose was 20 mg/day and it was increased by 20 mg every 2 days to a target dose of 120–160 mg/day (weight ≥45 kg) or 60–80 mg/day (≤45 kg). After reaching the target dose, ziprasidone could be flexibly dosed at 80–160 mg/day (40–80 mg/day for subjects ≤45 kg).
Ziprasidone failed to separate from placebo in the treatment of schizophrenia in adolescents and both studies were terminated after the randomized controlled trial (RCT) demonstrated futility. The failure of the RCT to demonstrate ziprasidone's efficacy over placebo may have been due to the higher placebo response observed in some countries, which accounted for 27% of the study population. The most common treatment-emergent adverse events (≥10%) during the RCT were somnolence and extrapyramidal disorders, and somnolence only during the OLE. Overall, ziprasidone was often well tolerated with an overall neutral weight and metabolic profile.
Bipolar manic or mixed
Findling et al. (2013a) assessed the short- and long-term efficacy and safety of ziprasidone in children and adolescents 10–17 years of age with a manic or mixed episode associated with BP-I disorder in a 4-week, double-blind, multicenter, randomized placebo-controlled trial followed by a 26-week OLE. Subjects were randomized 2:1 to initially receive flexible-dose ziprasidone (40–160 mg/day) or placebo. Ziprasidone was administered with meals and titrated over a 1–2 week period from a starting dose of 20 mg/day up to a target dose of 120–160 mg/day (≥45 kg) or 60–80 mg/day (≤45 kg) by day 14. Following titration, further dosing adjustments were made if necessary within the range of 80–160 mg/day (subjects ≥45 kg), or between 40 and 80 mg/day (≥45 kg). The estimated least squares mean changes in YMRS total were −13.83 (ziprasidone) and −8.61 (placebo; p = 0.0005) at RCT endpoint.
In the OLE, 162 subjects were enrolled and the median duration of treatment was 98 days. The mean change in YMRS score from the end of the RCT to the end of the OLE was −3.3. Posthoc efficacy analyses for the RCT took into consideration the presence of key mania symptoms (grandiosity or elation/euphoria), parental history of bipolar disorder, and ADHD comorbidity, and ziprasidone was efficacious when compared to placebo in all of these analyses. The most common adverse events in the ziprasidone group during the RCT were sedation, somnolence, headache, fatigue, and nausea. Subjects in the OLE also presented with insomnia. One subject on ziprasidone in the RCT had Fridericia-corrected QT interval (QTcF) ≥460 mseconds. Elevated blood prolactin occurred more frequently in the ziprasidone group, while the incidence of all other frequently occurring abnormal laboratory findings (>10% in any group) was either similar in both groups or higher in the placebo group.
There were challenges in the conduct of this multisite study, which included dosing errors at three of the trial sites. It is possible that as a result of these challenges, ziprasidone was not FDA approved for the treatment of bipolar mania in pediatric patients, despite its observed positive effect for manic or mixed states. Another multisite RCT of a similar study design examining ziprasidone's acute efficacy and tolerability in pediatric BP-I disorder is currently ongoing (NCT02075047; clinicaltrials.gov).
Paliperidone
Paliperidone extended release (ER) currently has FDA approval for the treatment of adolescents 12–17 years of age with schizophrenia (Singh et al. 2011) and comes with weight-based dosing recommendations, specifically a starting dose of 3 mg/day with subsequent dosing between 3 and 6 mg/day for adolescents <51 kg and 3–12 mg/day for those ≥51 kg (Younis et al. 2013). Paliperidone is an active metabolite of its parent compound risperidone (Harrington and English 2010) and the ER formulation provides consistent plasma concentrations over 24 hours (Boom et al. 2009), allowing for once-daily dosing. It is eliminated primarily through renal excretion (Vermeir et al. 2008). See Table 7 for selected recent primary paliperidone intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
ABC-I, Aberrant Behavior Checklist-Irritability; CGI-I, Clinical Global Impressions-Improvement; DB, double blind; HDL, high-density lipoproteins; PANSS, Positive and Negative Syndrome Scale; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; YMRS, Young Mania Rating Scale.
Schizophrenia
Savitz et al. (2015a) directly compared the efficacy, safety, and tolerability of paliperidone ER in adolescents with schizophrenia to that of aripiprazole. This randomized double-blind trial assigned participants to either paliperidone ER (3–9 mg/day, flexible dosing) or aripiprazole (5–15 mg/day, flexible dosing) for an 8-week double-blind acute treatment period followed by an 18-week double-blind maintenance phase. The study was completed by 76% of participants (75% of paliperidone ER group and 77% of aripiprazole group) and demonstrated no significant difference in PANSS total scores between the two treatment groups, with both sets of patients demonstrating clinically meaningful improvement at endpoint (day 56) and a continued decrease in scores up to day 182. Adverse events, including weight gain, were observed more frequently with paliperidone ER (77% vs. 66.7%). It was concluded that both medications provided clinically meaningful symptomatic and functional improvement in this population.
Savitz et al. (2015b) also investigated the medication's long-term safety in adolescents with schizophrenia in a 2-year open-label study. Patients 12–17 years of age were flexibly dosed between 1.5 and 12 mg/day and, after completion of the study by 220 participants (184 completed the 2 years, 36 completed a 6-month study before trial extension), the results showed an improvement in schizophrenia symptoms (as measured by PANSS total score) within the first 3 months. Patients were generally able to sustain improvement until the endpoint with 41.7% achieving remission, thus supporting the clinical recommendation for long-term antipsychotic treatment. The safety profile was consistent with established information regarding paliperidone ER in adults and risperidone in adolescents, although it was additionally noted that patients ≥51 kg had a higher incidence of adverse events compared to those <51 kg (88.0% vs. 77.2%). The authors concluded that paliperidone demonstrated efficacy and general tolerability as a maintenance treatment option in this population.
Asenapine
Asenapine is a second-generation atypical antipsychotic administered sublingually. In the United States, asenapine was approved in adults as monotherapy in 2009 and as adjunctive therapy with lithium or valproate in 2010 for manic or mixed episodes associated with BP-I disorder. Asenapine became FDA approved in 2015 for patients aged 10–17 years with an acute manic or mixed episode associated with BP-I disorder. The pharmacokinetic profile of asenapine is similar among adolescents and adults. In the pediatric population, asenapine is rapidly absorbed with maximum plasma concentrations achieved within 1.5 hours. Asenapine exposure increases in a dose-proportional manner over the range of 1–10 mg. Clearance and volume of distribution are linear with respect to time and dose. Steady state is achieved within 6–8 days of twice-daily (BID) dosing, consistent with a terminal elimination half-life of ∼20 hours (Findling et al. 2015c). The pharmacologic profile of asenapine is characterized by high affinity and antagonism of serotonergic receptors and potent dopamine, adrenergic, and histamine antagonism with low cholinergic activity (McIntyre and Wong 2012). See Table 8 for selected recent primary asenapine intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
BID, bis in die (twice a day); BP-I, bipolar I disorder; OLE, open-label extension; PANSS, Positive and Negative Syndrome Scale; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial; RPCT, Randomized Placebo-Controlled Trial; SD, standard deviation; YMRS, Young Mania Rating Scale.
BP-I manic/mixed
Findling et al. (2015c) studied adolescents 10–17 years of age with BP-I disorder currently in manic or mixed episodes in a 3-week double-blind, randomized placebo-controlled trial, with patients randomized 1:1:1:1 to placebo, asenapine 2.5, 5, or 10 mg BID. The starting dose was 2.5 mg BID, which was increased every 3 days to the dose target for the group. All asenapine doses were superior based on change in YMRS, with significantly higher 50% YMRS responder rates (42%–54%) compared to placebo (28%) on day 21. Asenapine was generally well tolerated. There was a higher incidence of somnolence, sedation, and hypersomnia in the asenapine treatment groups versus placebo. There was also a higher incidence of oral hypoesthesia with dysgeusia in the treatment groups versus placebo. Oral hypoesthesia and paresthesia are unique to asenapine and result from the local anesthetic properties of the sublingual formulation. The asenapine groups also had a higher incidence of weight gain and fasting insulin, cholesterol, triglycerides, low-density lipoprotein, and glucose changes than the placebo group.
Findling et al. (2016) conducted the first long-term safety and tolerability study with patients 10–17 years of age with an acute manic or mixed episode associated with BP-I disorder. Following the above-mentioned study (Findling et al. 2015c), patients could enroll in this flexible-dose (2.5–10 mg BID) OLE study for an additional 50 weeks. Open-label asenapine was started at 2.5 mg BID on day 1 and increased to 5 mg BID on day 4. At the day 7 visit, the asenapine dose was increased to 10 mg BID if tolerated, after which asenapine dosing was flexible based upon tolerability and/or symptomatology.
In terms of efficacy, the improvement in mania as measured by the YMRS total score from baseline was maintained over the course of the extension trial. In addition, patients who transitioned to asenapine in the OLE after receiving placebo during the acute phase achieved the same level of response as the asenapine/asenapine group by the end of the extension study. After 26 weeks of open-label asenapine treatment, the majority of patients achieved clinically meaningful symptom improvement.
Long-term flexible dose administration of sublingual asenapine was generally well tolerated. Somnolence, weight gain, sedation, and headache were the adverse effects most commonly reported. Among the predefined treatment-emergent adverse events of interest, the combination of somnolence, sedation, and hypersomnia was most frequent (42.4% of the total treatment group) followed by oral hypoesthesia/dysgeusia, EPS, and dizziness. Significant weight gain (≥7% increase) was experienced by 34.8% of the patients. Even though asenapine was overall well tolerated, 56.4% of patients discontinued from the total number, 15% due to adverse effects.
Schizophrenia
Findling et al. (2015b) evaluated the safety and efficacy of asenapine in adolescents 12–17 years of age, who met DSM-IV-TR criteria for schizophrenia in an 8-week randomized placebo-controlled trial that randomized 1:1:1 to placebo, asenapine 2.5 mg BID, and asenapine 5 mg BID, plus a 26-week OLE with flexible dosing of asenapine. The starting dose was 2.5 mg BID with an increase to 5 mg BID on day 4 for those in the 5 mg BID group. In the OLE, the participants started on day 1 with asenapine 2.5 mg BID, and on day 4, the dose was increased to 5 mg BID. From day 5, dosing was adjusted to 2.5 or 5 mg BID based upon tolerability and/or symptomatology. The mean average dose was 8.7 mg/day.
Mean differences between asenapine and placebo on the PANSS total score on day 56 were not significant. Significant improvement in the CGI-S score was observed in the 5 mg BID group vs placebo on day 56. In the OLE, PANSS total scores decreased by −16.1 points in the group previously treated with placebo and −11.2 points in the continuous asenapine group from OLE baseline to week 26. In the acute phase, weight gain and the composite event of somnolence, sedation, and hypersomnia were more common in both asenapine groups than in the placebo group. Akathisia, fasting glucose elevation, and extrapyramidal syndrome were also more common in the 5 mg BID group than in the placebo group. There were no unexpected adverse events in the OLE. The mean change from baseline to endpoint of fasting insulin was greater for the 2.5 and 5 mg BID doses of asenapine compared to placebo, with a dose–response for asenapine.
Iloperidone
As with other second-generation antipsychotics, iloperidone acts through a combination of dopamine D2 and serotonin 5-HT2 antagonism (Citrome 2010; Meltzer and Massey 2011). It is approved for the treatment of schizophrenia in adults, but has no FDA-approved indications in children. Iloperidone has high binding affinity to serotonin 5-HT2A, dopamine D2 and D3 (Kalkman et al. 2003), and norepinephrine NE α-1 receptors (Richelson and Souder 2000). Its elimination is through hepatic metabolism involving CYP2D6 and CYP3A4 isozymes of the cytochrome P450 group. Iloperidone is associated with QTc interval prolongation comparable to ziprasidone and haloperidol (Caccia et al. 2010) in adults with schizophrenia. No published prospective studies were found in children and adolescents over the past 5 years.
Lurasidone
Lurasidone has FDA approval for adolescents with schizophrenia (13–17 years) and very recently received an indication for BP-I depression (10–17 years). It is characterized by high-affinity binding to D2 (antagonist), 5-HT2A (antagonist), 5-HT7 (antagonist), and 5-HT1A (partial agonist) receptors, but has also demonstrated moderate affinity for noradrenergic α2C and α2A receptors, and a weak affinity for 5-HT2C receptors (Ishibashi et al. 2010). It is recommended that lurasidone be taken with at least a 350-Kcal meal as Cmax and AUC were found to be substantially higher (threefold and twofold, respectively) when administered with food (Preskorn et al. 2013).
When looking at the pharmacokinetic profile of lurasidone in children (6–17 years of age) dosed between 20 and 160 mg/day, Findling et al. (2015a) found general similarity to that seen in adults with the exception of slightly higher exposure levels seen in younger children at some doses (e.g., 120 mg). A linear dose effect on drug exposure was observed on day 1 (20–80 mg/day) and at steady state (20–160 mg/day) on day 10/12, with similar linear effects seen for all three active metabolites.
The adverse event profile demonstrated a higher frequency of somnolence in the study sample when compared with adults, but few adverse events related to movement disorders. Patients at higher doses (120 and 160 mg/day), especially younger participants, were found to have a higher likelihood of experiencing an adverse event, and it was concluded that a dose range of 20–80 mg/day provided adequate serum concentrations along with more acceptable tolerability. For that reason, this dose range was subsequently used in several key studies of lurasidone in youths. See table 9 for selected recent primary lurasidone intervention trials in pediatric populations.
Effect sizes are reported as Cohen's d.
Percentages reflect adverse events observed in active medication/treatment groups.
ABC-I, Aberrant Behavior Checklist-Irritability; ASD, autism spectrum disorder; CDRS-R, Children's Depression Rating Scale-Revised; PANSS, Positive and Negative Syndrome Scale; RDBPCT, Randomized Double-Blind Placebo-Controlled Trial.
Autism spectrum disorder
Lurasidone's potential as a treatment for irritability associated with ASD in children between 6 and 17 years of age was investigated by Loebel et al. (2016) who, after 6 weeks of treatment with either fixed, once-daily doses of lurasidone (20 or 60 mg/day) or matching placebo, found no significant difference in endpoint irritability as measured by the ABC-I subscale scores. Although improvement by endpoint on the CGI-I score was significant for the lurasidone 20 mg/day group, with the 60 mg/day group showing numerical improvement, there were no significant differences observed on additional secondary efficacy measures, including other ABC subscales (hyperactivity, stereotypic behavior, inappropriate speech, and lethargy/withdrawal), the Children's Yale-Brown Obsessive Compulsive Scales (CY-BOCS) modified for pervasive developmental disorders, and the Caregiver Strain Questionnaire (CGSQ). It was concluded that the results of this study did not confirm the efficacy of lurasidone for this patient population.
Schizophrenia
Goldman et al. (2017) studied the efficacy and safety of lurasidone after 6 weeks in adolescents with schizophrenia, 13–17 years of age, and found that fixed-dose lurasidone (40 or 80 mg/day) brought about statistically significant and clinically meaningful improvement in symptoms (−18.6 and −18.3 in PANSS total score respectively) compared to placebo, with similar improvement in both the 13–15 and 16–17 year groups. The lurasidone groups also demonstrated significant improvement in clinical severity, functionality, and quality of life. The authors concluded that lurasidone was a safe and effective option for short-term treatment in this population.
Bipolar depression
A more recent study by DelBello et al. (2017) looked into the efficacy and safety of lurasidone in children and adolescents 10–17 years of age with BP-I depression. When flexibly dosed from 20 to 80 mg/day with mean daily doses of 31.5 mg/day in the 10–14-year-old group, 33.8 mg/day in the 15–17-year-old group, and 32.5 mg/day in the combined age groups, the lurasidone-treated patients demonstrated statistically significant and clinical meaningful improvement in depressive symptoms (CDRS-R total score mean change of −21.0 vs. −15.3 in placebo; effect size 0.45) after 6 weeks, in addition to significant improvement in anxiety, quality of life, and global functioning. Effect sizes were observed to be larger for the older participant group (0.68) compared to the younger (0.13), which was accounted for, in part, by the greater improvement on placebo in the younger group. It was concluded that lurasidone monotherapy significantly improved depressive symptoms in this pediatric population and was generally well tolerated.
Cariprazine
Another second-generation antipsychotic, cariprazine, acts through a combination of partial agonist activity with high binding affinity at central dopamine D2 and serotonin 5-HT1A receptors, and antagonist activity with high to moderate affinity at serotonin 5-HT2A receptors (Kiss et al. 2010; Agai-Csongor et al. 2012). It is approved for the treatment of schizophrenia and the treatment of acute manic or mixed episodes associated with BP-I disorder in adults, but has no FDA-approved indications in children (Federal Drug Administration 2015). Cariprazine has two major active metabolites, desmethyl cariprazine and didesmethyl cariprazine, which are pharmacologically equipotent to cariprazine. It is metabolized by CYP3A4 and to a lesser extent by CYP2D6 (Kirschner et al. 2008). Cariprazine should not be given concomitantly with CYP3A4 inducers and its dosing should be reduced by half when co-administered with strong CYP3A4 inhibitors.
Cariprazine's half-life is 2–6 days, while one of its metabolites (didesmethyl cariprazine) has a half-life of 14–21 days. Due to its long half-life, patients need to be monitored for treatment response and late-occurring adverse reactions for several weeks after starting and with each dosage change. Cariprazine is generally well tolerated when given to adults with schizophrenia and bipolar disorders. It is associated with adverse effects such as insomnia, EPS, sedation, akathisia, nausea, dizziness, vomiting, and constipation when given to adults with schizophrenia (Citrome 2013a, 2013b). No published prospective studies were found in children and adolescents over the past 5 years.
Clinical Significance
Antipsychotics are commonly prescribed medications for children and adolescents. Historically, there has been a paucity of data on the efficacy and tolerability of these agents in pediatric populations. However, as shown in this review, a number of methodologically rigorous clinical trials have been completed over the past 5 years, the results of which can meaningfully inform clinical practice. An ever-increasing body of knowledge is now available for the clinical practitioner considering the use of an antipsychotic agent as treatment in a child or adolescent. This review of recent clinical trials shows that more data are available in regard to both the acute efficacy and, to a lesser extent, the postacute efficacy and tolerability for several of the considered antipsychotic medications. Conditions that have been studied include monotherapy in schizophrenia, bipolar disorder, and autism, as well as adjunctive treatment of DBDs with aggression.
The atypical antipsychotic class of medications has more recently been used preferentially over the typicals for a variety of clinical situations due to their putative reduced propensity to cause EPS when compared to first-generation “neuroleptics.” Despite the concerns for weight gain and obesity-related health outcomes, such as cardiovascular disease and diabetes, the incidence of dyskinesias and other EPS tends to be comparatively lower for the newer agents.
The atypical antipsychotics have been shown to be effective and generally well tolerated when used to treat psychotic symptoms, although their utility in treating symptoms of prodromal schizophrenia in at-risk patients is less clear (McGorry et al. 2013). For the treatment of nonpsychotic disorders, these medications are frequently used as adjunct treatment in disruptive behaviors, especially for ADHD with aggression not responding to optimized doses of stimulants, and for the irritability and behavioral problems frequently seen with ASD. Specific agents are usually selected for a given patient population based upon what is known about their efficacy and tolerability. When considering the durability of the therapeutic effect and how long these medications should continue to be administered, there are many issues relating to their impact on maturation, physical growth, and possible development of reduced effectiveness that are relevant for long-term efficacy (Lemmon et al. 2011; Marcus et al. 2011).
Some of the limitations discussed in the above studies include relatively small sample sizes, lack of comorbidity inclusions, short-term use of medication, and use of concomitant medications. The generalizability of findings to other clinical situations is therefore restricted and, given that most studies do not extend beyond a few months or a year, there is often little available information regarding the long-term effects of these medications. For some antipsychotic agents, despite being approved for use in children, the tested dosage ranges are frequently lower than what is prescribed in the adult population, and the effects at higher dosages have yet to be determined.
When considering the direction of future research, there is a need for studies that evaluate expanded treatment lengths, dosage, co-administration with other agents, and age. The lack of psychopharmacological studies in very young children limits the treatment options available to that population. There is sometimes reticence in families to enroll their children and adolescents in clinical trials, and longer-term studies may selectively involve families with a substantive commitment to study participation. This, in turn, may result in a possible bias toward more positive outcomes (Findling et al. 2017a).
Another consideration is the difficulty of diagnostic precision, particularly in early presentations of psychosis or mood disturbance. Furthermore, there are few methodologically rigorous blinded, head-to-head studies involving active controls available to inform medication selection for specific individual patients. In addition to the need for longer-term studies to assess efficacy and safety as well as remission and relapse rates, it may be that future research will identify a means by which medications can be selected in a more patient-specific way. Considering that aggression is oftentimes a key target symptom for which antipsychotics are prescribed (USDHHS 2009), the role of these medications in addressing dysfunctional and maladaptive aggression within the context of DBDs is one such area that warrants further study.
Conclusion
In summary, although evidence is mounting regarding the efficacy and tolerability of antipsychotic medications for mental health disorders in children and adolescents, more research is needed so that evidence-based comparisons between medications can be made in patients across developmental status, psychiatric comorbidities, and all socioeconomic groups. The atypical antipsychotics have shown tremendous potential in treating myriad psychiatric illnesses in younger patients, and the functional knowledge pertaining to this diverse class of medications will only grow in the upcoming years. It is to be hoped that future research will better enable informed treatment choices that take into account individual patient characteristics when selecting psychopharmacological agents, and that these data can be incorporated into evidence-based practices in clinical settings.
Footnotes
Disclosures
Dr. Findling receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Aevi, Akili, Alcobra, Amerex, American Academy of Child & Adolescent Psychiatry, American Psychiatric Press, Bracket, Epharma Solutions, Forest, Genentech, Guilford Press, Ironshore, Johns Hopkins University Press, KemPharm, Lundbeck, Merck, NIH, Neurim, Nuvelution, Otsuka, PCORI, Pfizer, Physicians Postgraduate Press, Purdue, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, Syneurx, Teva, Tris, TouchPoint, Validus, and WebMD.
Dr. Lee and Dr. Vidal have no competing financial interests.