
Abstract
Objective:
Dasotraline is a potent inhibitor of presynaptic dopamine and norepinephrine reuptake with a pharmacokinetic profile characterized by slow absorption and a long elimination half-life. The aim of this study was to evaluate the efficacy and safety of dasotraline in children with attention-deficit/hyperactivity disorder (ADHD).
Methods:
Children aged 6–12 years with a Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) diagnosis of ADHD were randomized to 6 weeks of double-blind once-daily treatment with dasotraline (2 or 4 mg) or placebo. The primary efficacy endpoint was change from baseline in the ADHD Rating Scale Version IV–Home Version (ADHD RS-IV HV) total score at week 6.
Results:
A total of 342 patients were randomized to dasotraline or placebo (mean age 9.1 years, 66.7% male). Treatment with dasotraline was associated with significant improvement at study endpoint in the ADHD RS-IV HV total score for the 4 mg/day dose versus placebo (−17.5 vs. −11.4; p < 0.001; effect size [ES], 0.48), but not for the 2 mg/day dose (−11.8 vs. −11.4; ns; ES, 0.03). A regression analysis confirmed a significant linear dose–response relationship for dasotraline. Significant improvement for dasotraline 4 mg/day dose versus placebo was also observed across the majority of secondary efficacy endpoints, including the Clinical Global Impression (CGI)–Severity score, the Conners Parent Rating Scale-Revised scale (CPRS-R) ADHD index score, and subscale measures of hyperactivity and inattentiveness. Discontinuation rates due to adverse events (AEs) were higher in the dasotraline 4 mg/day group (12.2%) compared with the 2 mg/day group (6.3%) and placebo (1.7%). The most frequent AEs associated with dasotraline were insomnia, decreased appetite, decreased weight, and irritability. Psychosis-related symptoms were reported as AEs by 7/219 patients treated with dasotraline in this study. There were no serious AEs or clinically meaningful changes in blood pressure or heart rate on dasotraline.
Conclusion:
In this placebo-controlled study, treatment with dasotraline 4 mg/day significantly improved ADHD symptoms and behaviors, including attention and hyperactivity, in children aged 6–12 years. The most frequently reported AEs observed on dasotraline included insomnia, decreased appetite, decreased weight, and irritability.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder characterized by a persistent pattern of hyperactivity, impulsivity, and/or inattention associated with significant impairment in social, academic, or occupational functioning (American Psychiatric Association 2013). A pooled estimate, based on a meta-analysis of published studies, found the prevalence of ADHD in pediatric populations to be ∼7% (Thomas et al. 2015).
Onset of ADHD is typically in childhood, with an older age of onset reported in females (Davies 2014). Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) increased the maximum age-of-onset criteria from 7 to 12 years, which appear to have resulted in a moderate increase in illness prevalence (Vande Voort et al. 2014). ADHD is often chronic, with 40%–60% of childhood cases continuing into adulthood (Faraone et al. 2006; Spencer et al. 2007; Sibley et al. 2016).
Consistent with the high prevalence of ADHD, the National Ambulatory Medical Care survey reported an annual (2012–2013) average of 6.1 million physician office visits with ADHD as the primary diagnosis (pediatricians, 48%; psychiatrists, 36%; family practitioners, 12%), with a 2.5-fold higher visit rate for males (Albert et al. 2017). Stimulant medications were prescribed in 80% of visits, regardless of the age of the child.
Research has implicated monoaminergic receptor systems as neural substrates of ADHD (Gizer et al. 2009; Faraone and Mick 2010; Hawi et al. 2015; Gallo and Posner 2016). Consistent with this, drugs that target dopamine and norepinephrine receptor systems have demonstrated efficacy in the treatment of ADHD (May and Kratochvil 2010; Chan et al. 2016; Punja et al. 2016).
The most recent American Academy of Pediatrics clinical practice guideline (American Academy of Pediatrics 2011) recommends three classes of medication for the treatment of ADHD in children and adolescents, stimulants, norepinephrine reuptake inhibitors (atomoxetine), and selective α-2-adrenergic agonists (guanfacine, clonidine). Stimulants exhibit larger effect sizes (ESs) compared with atomoxetine and guanfacine (Faraone and Mick 2010; Bushe and Savill 2014; Rizzo and Martino 2015; Punja et al. 2016). However, approximately one-third of patients treated with stimulant medication do not respond adequately and/or experience safety or tolerability issues (Childress and Sallee 2014; Sikirica et al. 2015). Therefore, there is a clear need for additional classes of treatments for ADHD.
Dasotraline is a potent inhibitor of presynaptic dopamine (IC50 3 nM) and norepinephrine (IC50 4 nM) transporters (Hopkins et al. 2016). Unlike amphetamine and related compounds, dasotraline does not stimulate dopamine release. The pharmacokinetic (PK) profile of dasotraline in adults is characterized by slow absorption (t max, 10–12 hours) and a long elimination half-life (t ½, 47–77 hours) resulting in stable plasma concentrations over 24 hours with once-daily dosing (Chen et al. 2016). The PK profile of dasotraline in children and adolescents is similar to adults, with a t max of 9.6–12 hours and an elimination half-life of 56–84 hours (data-on-file). Unlike stimulants, dasotraline does not produce marked peak and trough effects, which may in part account for its low potential for abuse. Abuse potential of dasotraline has been investigated in a placebo-controlled, active-comparator (methylphenidate) abuse liability study in recreational stimulant users (Koblan et al. 2016) showing a reduced potential for abuse that was comparable to placebo. The pharmacological and pharmacokinetic profile of dasotraline makes it a promising candidate for the treatment of ADHD.
In a randomized, parallel-group placebo-controlled study of children with ADHD in a laboratory classroom setting (Goldman et al. 2018), once-daily evening treatment with dasotraline 4 mg was associated with significant improvement in ADHD measures, with an ES of 0.85 on the primary outcome, the Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP), a validated scale completed by trained observers that assesses the severity of behavioral manifestations of ADHD (Wigal et al. 1998). In addition, dasotraline, in doses of 8 mg/day, has demonstrated efficacy in adults with ADHD in a randomized, double-blind 4-week study (Koblan et al. 2015).
We report here the results of the first randomized, double-blind, parallel-group, 6-week study designed to evaluate the efficacy, safety, and tolerability of dasotraline for the treatment of ADHD in children aged 6–12 years.
Methods
In this multicenter outpatient study, children aged 6–12 years with a diagnosis of ADHD were randomized, double-blind, to receive 6 weeks of treatment with two fixed-doses of dasotraline (2 and 4 mg) or placebo, administered once-daily in the morning. Patients randomized to dasotraline 4 mg initially received 2 mg/day for the first 7 days before being increased to the 4 mg dose.
Randomization, in a 1:1:1 ratio, was performed using a computerized random number generator. An Interactive Response System was used to manage randomization at baseline. Study drug capsules were identical in appearance and packaging. Patients, relatives, research staff, and sponsor personnel were blinded to treatment allocation from the time of study initiation until the completion of data analysis. Before randomization, patients completed a 7- to 21-day medication washout.
The study was conducted at 40 sites in the United States between April 2015 and July 2016. The study was approved by the institutional review board at each investigational site and was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines and with the ethical principles of the Declaration of Helsinki. Written informed assent was obtained from the child, and written informed consent was obtained from at least one of the child's parents or legal guardians. A Data and Safety Monitoring Board reviewed safety data at regular intervals. Patients who completed treatment in this study were eligible to enroll in a 26-week open-label extension study (NCT02457819).
Study patients
Male and female children, aged 6–12 years, were eligible for enrollment who met the DSM-5 criteria for a primary diagnosis of ADHD, with an ADHD Rating Scale Version IV–Home Version (ADHD-RS-IV HV; modified for investigator administration; DuPaul et al. 1998) score ≥28 and Clinical Global Impression–Severity scale (CGI-S; Guy 1976) score ≥4. Diagnosis was confirmed at screening using the Kiddie Schedule for Affective Disorders and Schizophrenia for School-Age Children–Present and Lifetime Version (K-SADS-PL) (Kaufman et al. 1997).
Exclusion criteria included a diagnosis of bipolar or major depressive disorder, conduct disorder, obsessive-compulsive disorder, disruptive mood dysregulation disorder, intellectual disability, any history of psychosis, autism spectrum disorder, and Tourette syndrome; any Central Nervous System Disorder suicidal ideation; and any acute or unstable medical condition and/or clinically significant laboratory abnormalities.
Concurrent cognitive behavioral therapy was permitted if treatment was stable and was initiated at least 1 month before screening. Prohibited medications included antipsychotic medication (within 8 weeks before screening), lithium, alpha-2 adrenergic receptor agonists, sedating antihistamine medications, and any ADHD, antidepressant or anticonvulsant medication.
Efficacy and safety measures
The a priori primary efficacy endpoint was change from baseline to week 6 in ADHD symptoms as measured by the ADHD RS-IV HV total score. The ADHD RS-IV HV is an 18-item scale, linked to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) (American Psychiatric Association 1994) diagnostic criteria for ADHD, which rates individual items on a 4-point severity scale based on a semi-structured interview with the patient's parent or primary caretaker. Additional assessments included the ADHD RS-IV HV inattentiveness and hyperactivity subscales (DuPaul et al. 1998), the CGI-S (Guy 1976), and the Weekly Parent Ratings of Evening and Morning Behavior-Revised (WPREMB-R) (Wehmeier et al. 2009). Efficacy was also assessed using the full-length version of the Conners Parent Rating Scale-Revised scale (CPRS-R), which includes the ADHD index score, and inattention and hyperactivity/impulsivity subscale scores (Conners et al. 1998). The Conners Teacher Rating Scale-Revised scale (CTRS-R) was also included as an efficacy measure; however, results for the CTRS-R scale were uninterpretable due to small sample sizes (17% of intent-to-treat [ITT] population) and are not summarized in the current report. Functional outcome was assessed by the Weiss Functional Impairment Rating Scale–Parent Report (WFIRS-P) (Canadian Attention Deficit Hyperactivity Disorder Resource Alliance [CADDRA] 2011). Rater training was provided for all study assessment measures by an independent rater qualification service.
Safety endpoints included physical examinations, 12-lead electrocardiogram (ECG), vital signs, adverse event (AE) reports, clinical laboratory results, Children's Sleep Habits Questionnaire (CSHQ; Owens et al. 2000), Columbia Suicide Severity Rating Scale (C-SSRS) Children's Assessment (Posner et al. 2011), weight, and body mass index (BMI). Risk of psychosis-related events was assessed based on AE terms in a review for the FDA (PID No. D060163; as summarized in Mosholder et al. 2009).
Statistical analysis
The safety population consisted of all patients who were randomized and received at least one dose of study medication; and the ITT population consisted of patients in the safety population who had at least one post-baseline ADHD RS-IV HV assessment.
The primary efficacy endpoint (ADHD RS-IV HV) was analyzed using a mixed model for repeated measures (MMRM) in the ITT population. For the MMRM, no imputation for missing data was applied. The MMRM model included treatment, visit (as a categorical variable), pooled center, baseline ADHD RS-IV HV total score, and treatment-by-visit interaction. Secondary efficacy endpoints were evaluated, where applicable, using an MMRM analysis. Endpoint response, defined a priori as ≥30% decrease from baseline in the ADHD RS-IV HV total score, was analyzed using a logistic regression model, with baseline ADHD-RS-IV HV total score, pooled center, and treatment in the model. In the analysis of change from baseline in ADHD RS-IV HV total scores, a Hochberg testing procedure was used to control family-wise Type I error at 5%. Secondary efficacy analyses were not adjusted for multiplicity.
To explore the robustness of the MMRM results for the primary efficacy variable, sensitivity analyses were performed using a placebo-based multiple imputation pattern-mixture model and a tipping point analysis of the pattern-mixture model.
Preplanned MMRM analyses of efficacy were performed on the following clinical subgroups: age, gender, weight, and prior treatment with ADHD medication.
Based on the results from previous pediatric studies, we calculated that a sample size of 82 patients per treatment group was needed to provide 80% power to detect a treatment difference as significant at the 0.05 level (two-sided) between each dose of dasotraline and placebo. With an expected attrition rate of 25%, it was necessary to randomize at least 110 patients per treatment group.
Descriptive statistics were used for safety variables, including AEs, vital signs, and laboratory results. Number needed to harm (NNH) for selected AEs was calculated by assessing the reciprocal of the difference in adverse effect rates for the dasotraline and placebo groups. Week 6 change in BMI was expressed in terms of mean change the z-score. Cohen's d ESs were calculated for efficacy measures as the difference in drug versus placebo change scores divided by the pooled standard deviation, based on an MMRM analysis (Cohen 1992).
Results
Patients
A total of 593 patients were screened for inclusion, of whom 342 (57.7%) comprised the safety population, and 336 comprised the ITT population (Fig. 1). The three treatment groups were comparable in terms of clinical and demographic characteristics at baseline (Table 1).
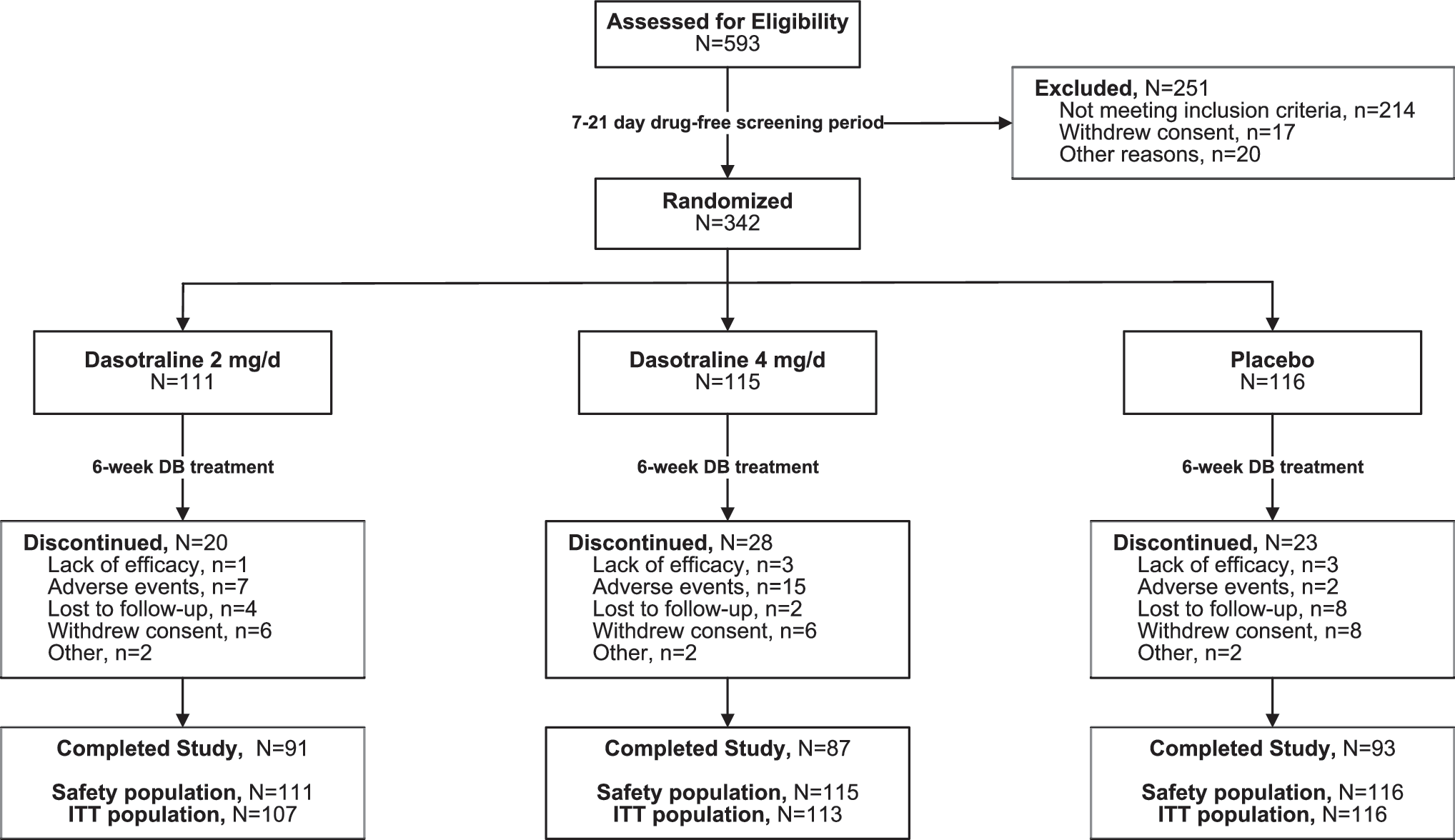
Study flow diagram.
Baseline Characteristics (Safety Population)
Incidence >5%.
ADHD Rating Scale Version IV–Home Version.
ADHD, attention-deficit/hyperactivity disorder; BMI, body mass index; CGI-S, Clinical Global Impression–Severity scale; ITT, intent-to-treat.
Study completion rates for dasotraline 2 mg/day, 4 mg/day, and placebo were 82.0%, 75.7%, and 80.2%, respectively (Fig. 1). Among study completers, 87.5% continued into a 26-week open-label continuation study.
Efficacy
Least squares mean change from baseline to week 6 was significantly greater in the dasotraline 4 mg/day group compared with the placebo group on the ADHD RS-IV HV total score (primary endpoint, −17.5 vs. −11.4; p < 0.001; ES: 0.48; Table 2). On secondary measures, least squares mean change at week 6 was significantly greater in the dasotraline 4 mg/day group compared with the placebo group on the CGI-S score (−1.4 vs. −1.0; p = 0.04; ES: 0.29) and CPRS-R index score (−28.9 vs. −18.2; p = 0.015; ES: 0.35). Treatment with dasotraline 2 mg/day was not significantly different from placebo on the primary ADHD RS-IV HV endpoint (−11.8 vs. −11.4; p = 0.81) or on secondary efficacy measures (Table 2).
Efficacy Endpoints: Change at Week 6 (Intent-to-Treat Population; Mixed Model for Repeated-Measures Analysis)
ADHD Rating Scale Version IV–Home Version.
ADHD, attention-deficit/hyperactivity disorder; CGI, Clinical Global Impression; CPRS-R, Conners Parent Rating Scale-Revised; LS, least squares; n.s., not significant.
For the 4 mg/day dose of dasotraline, the robustness of the primary efficacy analysis (endpoint change in ADHD RS-IV HV) was confirmed based on sensitivity analyses (pattern-mixture model and tipping point analyses). A significant linear dose–response relationship was demonstrated for dasotraline (p = 0.001; first-order regression analysis).
Significant improvement compared with placebo was observed starting at week 1 for dasotraline 4 mg/day on the ADHD RS-IV HV total score (Fig. 2) and the CGI-S score (Fig. 3) and was maintained at all subsequent study visits. Significant improvement was observed starting at week 3 on the CPRS-R ADHD index score.
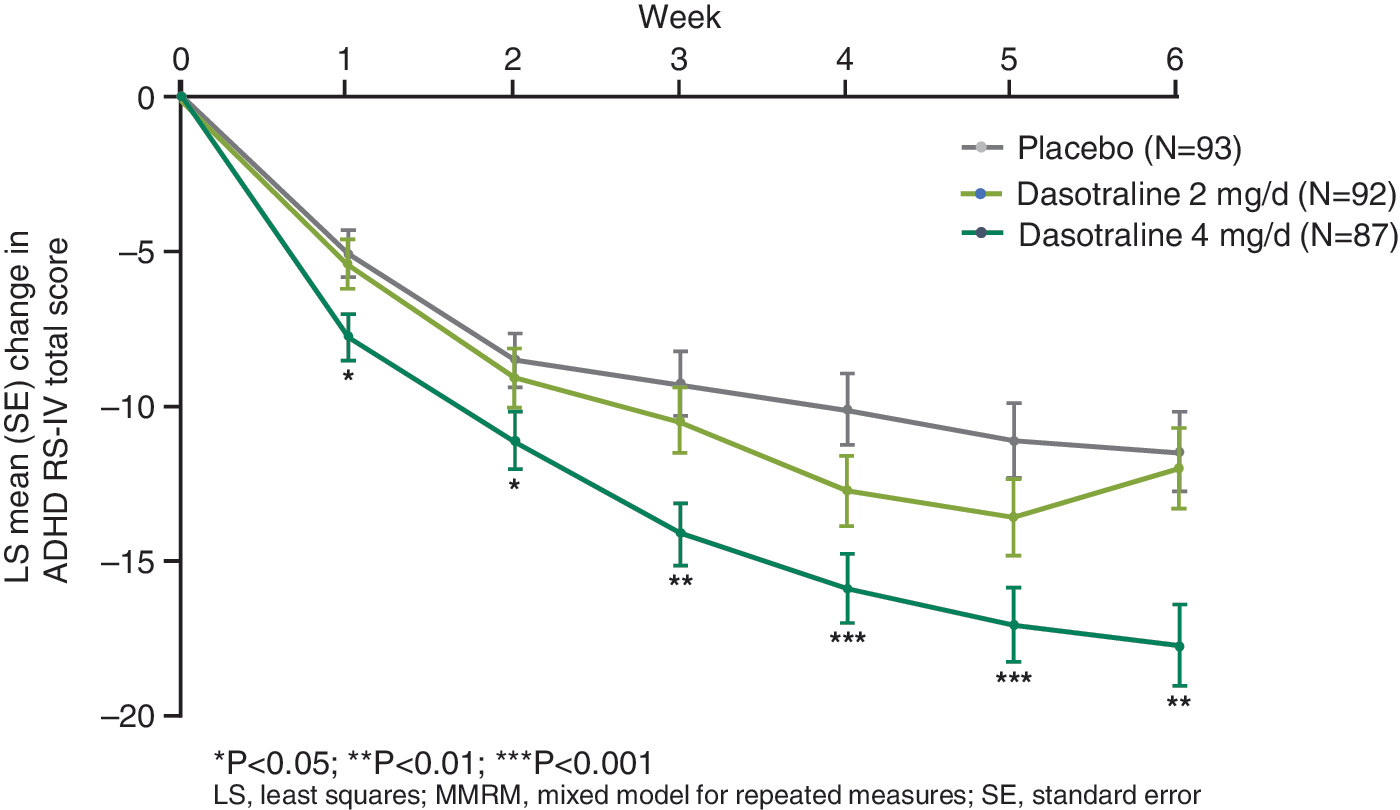
Least squares mean (SE) change from baseline in the ADHD RS-IV total score (ITT population, MMRM). ADHD RS-IV, attention-deficit/hyperactivity disorder Rating Scale Version IV; ITT, intent-to-treat; MMRM, mixed model for repeated measures.
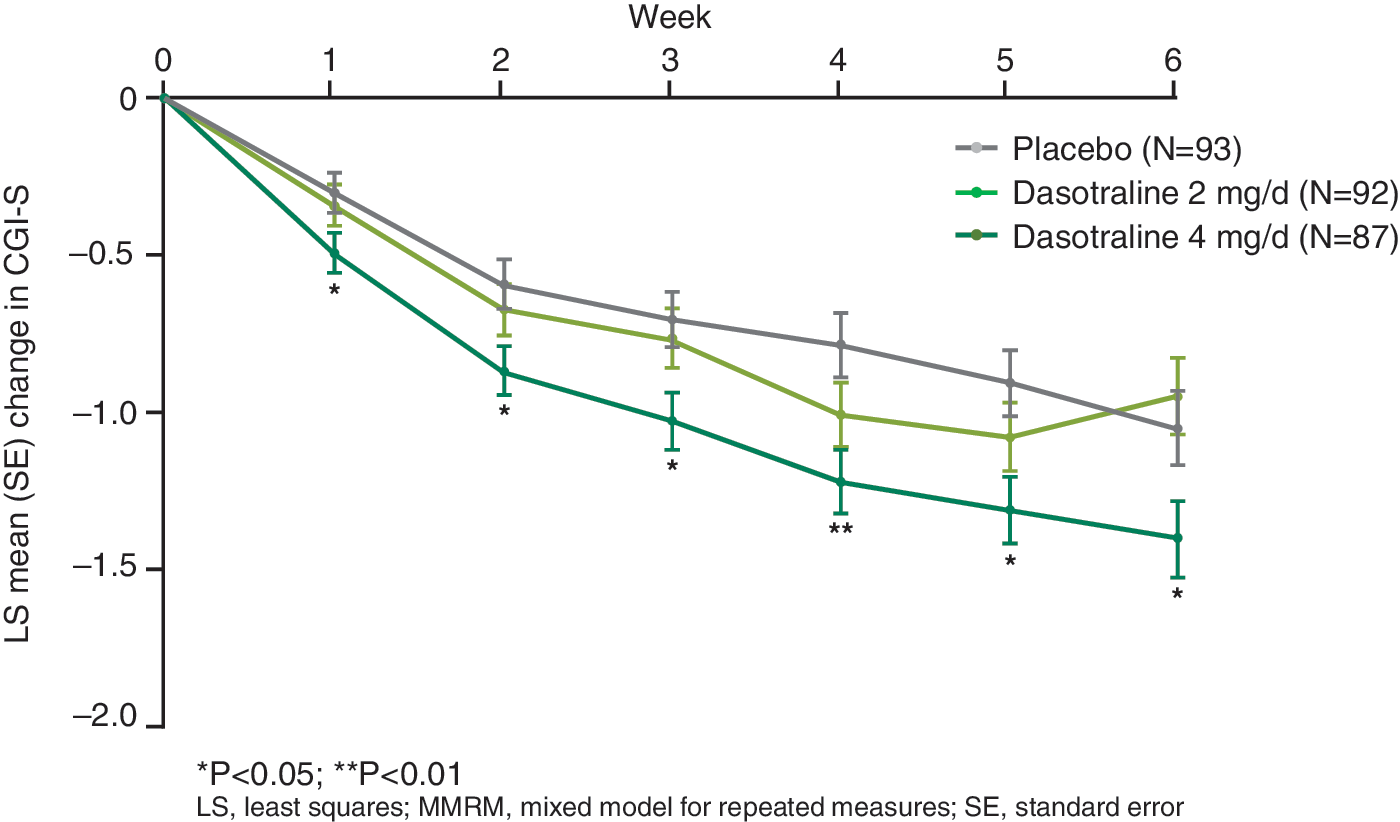
Least squares mean (SE) change from baseline in CGI-S score (ITT population, MMRM). CGI-S, Clinical Global Impression–Severity; ITT, intent-to-treat; MMRM, mixed model for repeated measures.
Treatment with dasotraline 4 mg/day (but not 2 mg/day) was associated with significant improvement at week 6 on the ADHD RS-IV HV inattentiveness and hyperactivity subscale scores (Table 2), with significant separation from placebo observed from week 1 onward. Significant improvement at week 6 was also observed on the CPRS-R inattention and hyperactivity/impulsivity subscale scores, with significant separation from placebo observed from week 4 onward for inattention and from week 3 onward for hyperactivity/impulsivity.
The proportion of responders (defined as ≥30% reduction in the ADHD RS-IV HV total score from baseline to week 6 last observation carried forward (LOCF) endpoint) was significantly higher in the 4 mg/day dasotraline group versus the placebo group (57.5% vs. 36.5%; NNT = 5; odds ratio 2.4, 95% confidence interval [95% CI] 1.38–4.01; p = 0.002; LOCF data; Fig. 4).
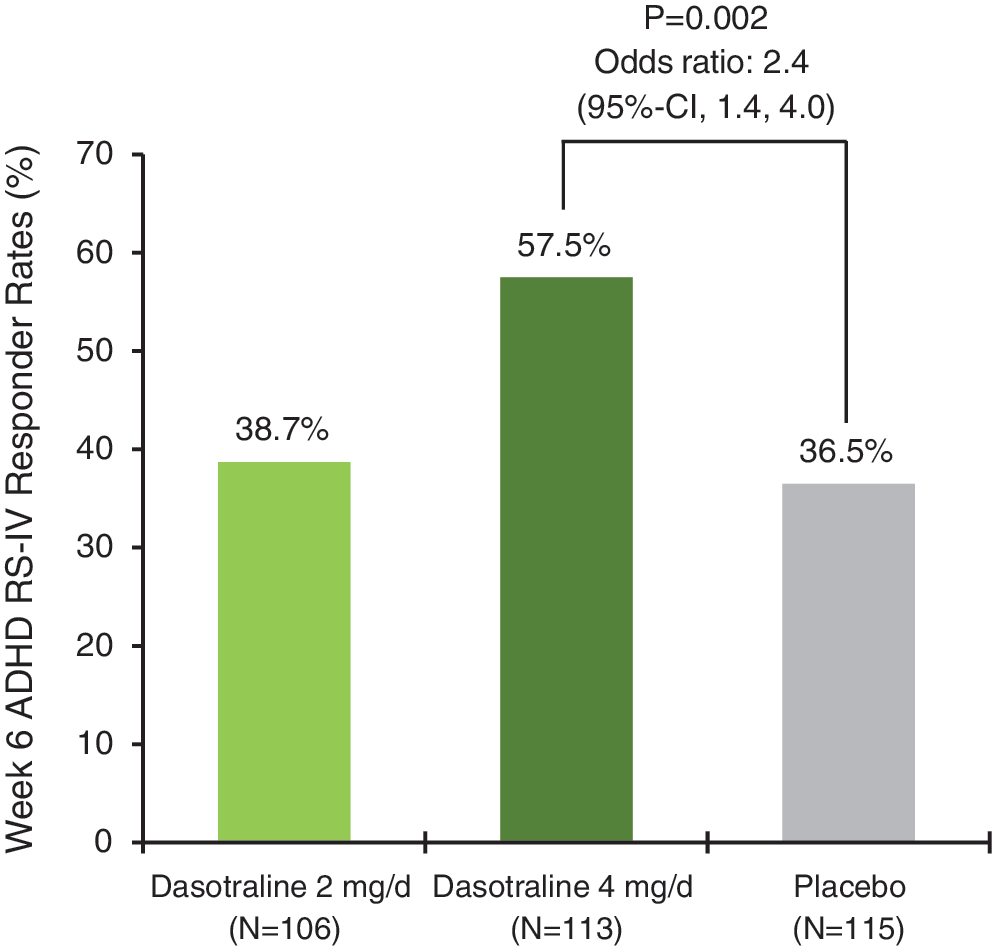
ADHD RS-IV HV responder rates at week 6 (LOCF; ≥30% reduction from baseline). ADHD RS-IV HV, attention-deficit/hyperactivity disorder Rating Scale Version IV–Home Version; LOCF, last observation carried forward.
No significant difference in improvement was observed on the functional outcome measure, the WFIRS-P, after 6 weeks of treatment with dasotraline 2 and 4 mg/day versus placebo (−0.21 and −0.21 vs. −0.17). Endpoint change was also not significant for either dose of dasotraline on the WPREMB-R total score.
Subgroup analyses
In a series of preplanned analyses of the effect of study treatment on the ADHD RS-IV HV total score, treatment with dasotraline 4 mg/day was associated with a larger endpoint ES in younger (6–9 years) versus older (10–12 years) patients (0.53 vs. 0.41; MMRM). ESs on the 4 mg/day dose of dasotraline were larger for male versus female patients (0.49 vs. 0.37; MMRM). Interaction tests (treatment-by-gender, treatment-by-age group) were not significant.
In patients (39.8%) reporting recent drug treatment for ADHD (94% stimulants), the ES at endpoint of dasotraline 4 mg/day was smaller compared with patients receiving no recent treatment (0.47 vs. 0.59; MMRM). Finally, in a post hoc analysis, patients in the dasotraline 2 mg/day dose group who had a baseline weight ≤30 kg had a larger dasotraline vs. placebo ES for week 6 change in the ADHD RS-IV total score (0.36) compared with patients with a baseline weight >30 kg (0.24).
Safety
Adverse events
At least one treatment-emergent AE was reported by 57.1% of patients in the combined dasotraline dose groups and 46.6% in the placebo group. For events with a higher incidence on dasotraline compared with placebo, the three most frequent AEs in the dasotraline 2 and 4 mg/day groups (vs. placebo) were insomnia (15.3% [NNH = 10] and 21.7% [NNH = 6] vs. 4.3%), decreased appetite (12.6% [NNH = 14] and 21.7% [NNH = 7] vs. 5.2%), and weight decreased (5.4% [NNH = 19] and 8.7% [NNH = 12] vs. 0%; Table 3). The most frequently reported subtype of insomnia was initial insomnia, which occurred in the dasotraline 2 mg/day, 4 mg/day, and placebo groups at rates of 9.0%, 16.5%, and 1.7%, respectively. Of note, endpoint improvement was observed for the dasotraline 2 and 4 mg/day dose groups, respectively, in mean (95% CI) CSHQ total sleep disturbances score from week 1 (−1.4 [−2.4 to −0.4] and −0.6 [−1.7 to +0.5]) to week 6 (−3.9 [−4.9 to −2.8] and −3.8 [−5.1 to −2.5]). Improvement was also observed on the placebo group at week 1 (−2.7 [−3.7 to −1.6]) and week 6 (−5.0 [−6.5 to −3.6]).
Adverse Events and Changes in Weight and Vital Signs at Week 6 (Last Observation Carried Forward Endpoint; Safety Population)
Incidence ≥2% in either dasotraline group and incidence for either dasotraline group >placebo.
Combined insomnia (initial, middle, terminal).
Systolic blood pressure ≥20 mmHg decrease from supine to standing or diastolic blood pressure ≥10 mmHg decrease from supine to standing.
≥20 bpm increase from supine to standing.
Seven patients (6.3%) discontinued due to a treatment-emergent AE in the dasotraline 2 mg/day group (two patients with insomnia and one patient each with phobia, decreased appetite, aggression, syncope, and prolonged QTcF of 494 msec, with an increase from baseline <60 msec). Fourteen patients (12.2%) discontinued due to an AE in the dasotraline 4 mg/day group (four patients with insomnia and one patient each with irritability, abnormal behavior, ADHD, emotional poverty; visual, mixed, or hypnopompic hallucinations; chest pain, costochondritis, and pruritus) and two patients (1.7%) in the placebo group (irritability, increased hepatic enzyme).
The number of patients reporting an AE as “severe” was similar on dasotraline 2 mg/day (n = 1 patient reporting insomnia), dasotraline 4 mg/day (n = 3 patients reporting decreased appetite, anxiety, irritability, insomnia), and placebo (n = 2 patients reporting irritability and bone contusion). No deaths or serious AEs occurred in any of the three treatment groups.
Psychosis-related symptoms (e.g., hallucinations, illusions) were reported as AEs by seven patients treated with dasotraline in this study. The events were predominantly transient (resolving the same day in three patients, despite continuing dasotraline), mild or moderate in intensity, and occurring in younger patients with lower body weights—6 of 7 patients were aged 6–8 years and were receiving a weight-adjusted dose of ∼0.15 mg/kg or higher (Supplementary Table S1). The incidence of psychosis-related events in patients with body weight of ≥30 kg was 0.84% (1/119). No mania- or hypomania-related events were reported by any patients in this study.
On the C-SSRS, two patients in the placebo group reported treatment-emergent suicidal ideation, and one patient each in the dasotraline 2 and 4 mg/day dose groups reported passive ideation. These latter two patients also reported transient suicidal ideation as an AE; treatment was continued, and each event resolved the same day. There were no deaths, and no suicide attempts in the study.
Weight and BMI
The mean weight and BMI at baseline were similar for all the three treatment groups (Table 1). Similar mean changes at week 6 were observed for weight in the dasotraline 2 and 4 mg/day group versus placebo (−0.4 and −1.2 kg vs. +1.0 kg; LOCF endpoint; Table 3). Mean week 6 changes in BMI z-scores were −0.24 and −0.43 versus +0.11 for dasotraline 2 mg/day, 4 mg/day, and placebo, respectively.
Laboratory, vital signs, and ECG
Minimal mean changes (<3 mmHg) were observed in supine or standing systolic and diastolic blood pressure in the dasotraline and placebo treatment groups (Table 3). Comparable rates of orthostatic hypotension were observed for the dasotraline and placebo groups. There was a small dose-related increase in orthostatic tachycardia in dasotraline compared with placebo (Table 3). There were no notable treatment-emergent differences between the combined dasotraline dose groups and placebo in laboratory or ECG parameters. Mean LOCF endpoint change, for dasotraline 2 mg/day, 4 mg/day, and placebo, respectively, was as follows for the PR interval (−1.2, −3.5, and +1.5 msec), the QRS duration (+0.9, +0.4, and −0.2 msec), and the QTcF interval (−0.1, −1.3, and −0.3 msec). One patient in the dasotraline 2 mg/day group, with a baseline QTcF of 455 msec, had a prolonged QTcF of 494 msec, with an increase from baseline <60 msec; no patients in the dasotraline 4 mg/day or placebo groups had a QTcF >460 msec or an increase in QTcF ≥60 msec.
Discussion
The results of this randomized, double-blind, placebo-controlled, study demonstrate the efficacy, safety, and tolerability of dasotraline, a novel dopamine-norepinephrine reuptake inhibitor, for the treatment of ADHD in children aged 6–12 years. Treatment with once-daily dasotraline 4 mg was associated with statistically significant and clinically meaningful improvement in the ADHD RS-IV HV total score (primary efficacy endpoint; p < 0.001). Improvement was also observed across most secondary efficacy endpoints, including the CGI-S score (p = 0.040), the CPRS-R ADHD index score (p = 0.015), and measures of hyperactivity/impulsivity and inattentiveness (on the ADHD RS-IV HV subscales, p = 0.038 and 0.074, respectively). No improvement was observed on the 2 mg/day dose of dasotraline for the primary ADHD RS-IV HV measure (p = 0.812) or for associated secondary efficacy endpoints.
The treatment ES for dasotraline 4 mg/day (vs. placebo) in the current study was 0.48 on the primary ADHD RS-IV HV assessment at week 6. Reliable efficacy comparisons with other marketed agents require data from head-to-head trials; however, we note that the improvement in ADHD symptoms reported here is comparable to that reported for atomoxetine, and somewhat smaller than has been reported for stimulant medication (Stuhec et al. 2015). We also note that dasotraline, in a fixed-dose of 4 mg (vs. placebo), was found to have an ES of 0.85 in a study of laboratory classroom performance in children aged 6–12 years with ADHD (Goldman et al. 2018).
In previous short-term pediatric trials of stimulant and nonstimulant medications in ADHD, improvement in the ADHD RS-IV total score in the placebo group ranged from −5.0 to −9.0 (Kratochvil et al. 2003; Muir and Perry 2010; Stuhec et al. 2015; Maneeton et al. 2015). These findings contrast with the current study, where endpoint change on placebo was relatively high (−11.4). It is possible that the magnitude of the improvement in the placebo group in the current study may have reduced the overall dasotraline treatment effect. Reasons for increased placebo response in this study may include the use of more than one dasotraline treatment arm (a 2:1 ratio of dasotraline-to-placebo), the number and frequency of efficacy assessments, and patient age range in the study (Khan et al. 2003, 2004; Sanderson et al. 2013; Weimer et al. 2015).
In planned subgroup analyses of endpoint change in the ADHD RS-IV HV scale, no significant treatment interaction effect was observed for dasotraline 4 mg/day based on gender or age. The somewhat larger ES in the younger (6–9 years) vs. older (10–12 years) age groups (ES, 0.53 vs. 0.41) may have been due, in part, to weight-related differences in dasotraline exposure associated with younger versus older patients. Consistent with these findings, patients in the dasotraline 2 mg/day dose group with a baseline weight of ≤30 kg had a larger ES for week 6 change in the ADHD RS-IV total score compared with patients with a baseline weight of >30 kg (0.36 vs. 0.24). Therefore, it is possible that lower weight children (≤30 kg) could show clinically meaningful improvement on dasotraline dose of 2 mg/day. This question requires further investigation.
Treatment with dasotraline 4 mg/day was associated with greater improvement in ADHD symptoms in treatment-naive patients compared with those reporting previous treatment. This finding is consistent with the results from other ADHD studies (Stuhec et al. 2015), suggesting that the magnitude of symptom improvement in ADHD clinical trials may be influenced by prior treatment exposure. Further investigation is needed to more fully characterize the relationship between the efficacy of dasotraline, illness chronicity and previous treatment response.
Minimal improvement in functioning was observed after 6 weeks of treatment with dasotraline on the WFIRS-P. Results from previous short-term treatment studies have suggested that improvement in functioning in ADHD may lag behind symptomatic improvement (Buitelaar et al. 2009; De Bruyckere et al. 2016), which may, in part, explain the lack of improvement in functioning observed in the current trial.
The most frequently reported AEs associated with dasotraline were insomnia, irritability, and decreased appetite and weight, which are consistent with the central monoaminergic activity of dasotraline. In the dasotraline treatment groups, insomnia was rated as severe in 3 of 47 patients and was cited as the reason for discontinuation in 2 patients on the 2 mg/day dose and 4 patients on the 4 mg/day dose. However, we note that results from the CSHQ showed modest reduction in sleep disturbance for both doses of dasotraline.
In the current study, there were no deaths and no serious AEs on dasotraline, and no active suicidal ideation or attempts. Psychosis-related symptoms (hallucinations, illusions) were reported as AEs by seven patients treated with dasotraline. The events were rated as mild-to-moderate in severity, and were transient, resolving during continued treatment with dasotraline (n = 4 patients), or upon treatment discontinuation (n = 3). The events predominantly occurred in younger patients (aged 6–8 years) with lower body weight (weight-adjusted dasotraline dose of ∼0.15 mg/kg or higher). In the ≥30 kg patient subgroup, 1 of 119 patients (0.84%) reported psychosis-related symptoms. These findings suggest that, to minimize the risk of psychosis-related symptoms, treatment with the 4 mg/day dose of dasotraline should be limited to patients with body weight of at least 30 kg (yielding a weight-adjusted dose ≤0.15 mg/kg). In safety reviews of currently available stimulant and nonstimulant treatments for ADHD, the incidence of psychosis-related symptoms has been reported to range from 0% to 2.5% (Mosholder et al. 2009; Ramstad et al. 2018; Storebø et al. 2018). This suggests that weight-adjusted dasotraline dosing in patients 6-12 years is associatd with rates of psychosis-related sympotms that are similar to rates reported for currently available ADHD agents.
Minimal changes in heart rate and systolic and diastolic blood pressure (supine and standing) were observed during treatment with dasotraline; the changes were not clinically meaningful. No clinically meaningful changes in ECG parameters were observed.
Study limitations include the exclusion of most medical and psychiatric comorbidities, which may limit the generalizability of the findings; the absence of a treatment arm with an active comparator; and the relatively short duration of the trial. Secondary efficacy endpoint in this study were not adjusted for multiplicity, therefore these results should be interpreted with caution. In addition, tolerability may have been reduced in the higher dose treatment arm by utilization of a fixed-dose design, with a forced titration at 7 days to the 4 mg/day dose.
Conclusions
In this placebo-controlled fixed-dose study, treatment with dasotraline 4 mg/day significantly improved ADHD symptoms and behaviors, including attention and hyperactivity, in children aged 6–12 years. No significant improvement was observed overall for dasotraline 2 mg/day; however, further studies are needed to determine whether the 2 mg dose may have efficacy in lower weight children (<30 kg). The most common AEs reported on dasotraline were insomnia and decreased appetite, consistent with its central monoaminergic mechanism of action. Treatment with dasotraline 4 mg/day should be limited to children weighing at least 30 kg (with a weight-adjusted dose of ≤0.15 mg/kg) to minimize the risk of psychosis-related symptoms. The results of the current study provide efficacy and safety data indicating that dasotraline, dosed once-per-day, is a promising therapeutic option for the treatment of ADHD, with a reduced potential for abuse, and with a long elimination half-life permitting sustained efficacy across a 24 hour dosing interval.
Clinical Significance
Dasotraline is a potent dopamine and norepinephrine reuptake inhibitor, which, unlike amphetamines and related compounds, does not stimulate dopamine release. The PK profile of dasotraline in children is characterized by slow absorption and a long elimination half-life, resulting in stable plasma concentrations over 24 hours with once-daily dosing. The results of this randomized, double-blind, placebo-controlled 6-week trial demonstrate the efficacy, safety, and tolerability of once-daily dasotraline 4 mg for the treatment of ADHD in children aged 6–12 years. These findings suggest that dasotraline may be a potentially useful therapeutic alternative for the treatment of ADHD in children 6–12 years.
Footnotes
Acknowledgments
J.H. served as the statistical expert for this study. The authors thank Edward Schweizer, MD, of Paladin Consulting Group, who provided editorial and medical writing support under the direction of the authors, which was funded by Sunovion Pharmaceuticals. The authors also thank the patients and their families who participated in this study. Clinical Trial Registration:
Disclosures
R.L.F. receives or has received research support, acted as a consultant and/or served on a speaker's bureau for Aevi, Akili, Alcobra, Amerex, American Academy of Child & Adolescent Psychiatry, American Psychiatric Press, Bracket, Epharma Solutions, Forest, Genentech, Guilford Press, Ironshore, Johns Hopkins University Press, KemPharm, Lundbeck, Merck, NIH, Neurim, Nuvelution, Otsuka, Patient-Centered Outcomes Research Institute, Pfizer, Physicians Postgraduate Press, Purdue, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, Syneurx, Teva, Tris, TouchPoint, Validus, and WebMD. L.A.A. has received grant/research support from Sunovion Pharmaceuticals, Purdue Pharmaceuticals, Enzymotec, Shire Pharmaceuticals, and Lundbeck and has served as a consultant to Enzymotec, Alcobra Pharmaceuticals, Rhodes Pharmaceuticals, Shire Pharmaceuticals, the National Football League, and Major League Baseball. He has also received royalty payments (as inventor) from NYU for license of adult ADHD scales and training materials since 2004. L.A.A. has no conflicts in regard to stock ownership or speakers' bureaus. T.J.S. received research support or was a consultant from the following sources: Avekshan, Ironshore, Lundbeck, Shire Laboratories Inc, Sunovion, the FDA and the Department of Defense. Consultant fees are paid to the MGH Clinical Trials Network and not directly to T.J.S. T.J.S. received research support from Royalties and Licensing fees on copyrighted ADHD scales through MGH Corporate Sponsored Research and Licensing. Through MGH corporate licensing, T.J.S. has a US Patent (#14/027,676) for a non-stimulant treatment for ADHD and a patent pending (#61/233,686) for a method to prevent stimulant abuse. R.G., S.C.H., K.S.K., J.K., J.H., and A.L. are employees of Sunovion Pharmaceuticals, Inc., Marlborough, MA, and Fort Lee, NJ.
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.