
Abstract
Objective:
Cytochrome P4502C19 (CYP2C19) is a highly polymorphic gene that encodes an enzyme that metabolizes escitalopram and sertraline, two selective serotonin reuptake inhibitors (SSRIs) that are FDA approved for pediatric use and commonly used to treat anxiety and depressive disorders in youth. Using pharmacokinetic (PK) models in adolescents, we sought to (1) model SSRI dosing across CYP2C19 phenotypes to compare SSRI exposure (area under curve, AUC) and maximum concentration (C max), (2) evaluate the impact of b.i.d. dosing (in rapid metabolizers [RM] and ultrarapid metabolizers [UM]) on SSRI exposure and C max, and (3) determine pharmacogenomically-informed dosing strategies to provide similar exposure across CYP2C19 phenotypes in adolescents.
Methods:
Using PK parameters in CYP2C19 phenotype groups and previously reported pediatric PK data for escitalopram and sertraline, we modeled exposure (AUC 0–24) and C max and determined CYP2C19-guided dosing strategies.
Results:
Compared with normal CYP2C19 metabolizers treated with either escitalopram or sertraline, C max and AUC 0–24 were higher in slower metabolizers and lower in patients with increased CYP2C19 activity, although the magnitude of these differences was more pronounced for escitalopram than for sertraline. For escitalopram, poor metabolizers (PMs) require 10 mg/day and UMs require 30 mg/day to achieve an exposure that is equivalent to 20 mg/day in a normal metabolizer (NM). For sertraline, to achieve AUC 0–24 and C max similar to NMs receiving 150 mg/day, PMs require 100 mg/day, whereas a dose of 200 mg/day was required in rapid and UMs. For UMs, b.i.d. escitalopram dosing was necessary to achieve comparable trough levels and exposure to NMs.
Conclusions:
This simulation study raises the possibility that achieving similar escitalopram and sertraline plasma concentrations could require dose adjustments in CYP2C19 poor metabolizers and UMs, although the magnitude of these differences were more pronounced for escitalopram than for sertraline. However, prospective trials of pharmacogenomically guided dosing in the pediatric population are needed to extend the findings of these modeling studies.
Introduction
Cytochrome P450 2
Pharmacokinetic (PK) modeling incorporates individual patient characteristics to determine the exposure to a medication, including dose, body size, age, and the influence of genes that influence the metabolism of that medication. The exposure is approximated by the area under the concentration–time curve (AUC), and dose-limiting toxicities are often related to the maximum concentration (C max). Allometric scaling is used to account for differences in body size (Holford and Anderson 2017). PK models for citalopram and sertraline were developed before the knowledge of the influence of CYP2C19 on the metabolism of these medications, and most do not account for the variability in exposure introduced by the differences in activity of this enzyme. The studies that do account for the gene's influence on the PKs of these medications were performed in adults (Wang et al. 2001; Rudberg et al. 2008). Therefore, we have combined the allometric scaling to account for the body size of adolescents and the CYP2C19 metabolizing activity to demonstrate the magnitude of the influence of CYP2C19 in adolescents. In the context of therapeutic drug monitoring, PK modeling can be used to adjust exposure to achieve targeted concentration or exposure. However, in the absence of concentration data, the model approximates the exposure of the “average” patient of that age and size.
In adults with reduced CYP2C19 metabolism, plasma escitalopram concentrations are higher than those in patients with normal CYP2C19 metabolism—putting these patients at higher risk of SSRI-related side effects (Jukić et al. 2018). In addition, patients with faster CYP2C19 metabolism have lower plasma escitalopram concentrations and are at high risk of treatment failure compared with NMs (Hicks et al. 2015). In adults, the influence of CYP2C19 metabolizer status on escitalopram PKs is well described (Steere et al. 2015; Jukić et al. 2018); however, the relationship between these variants and efficacy in the pediatric population are less well understood (Ji et al. 2014).
For sertraline-treated adults, higher dose-adjusted sertraline (and its primary metabolite, N-desmethyl sertraline) concentrations were observed in patients with >1 allele encoding a defective CYP2C19 enzyme than NMs (CYP2C19 *1/*1) (Rudberg et al. 2008). In addition, among healthy Chinese volunteers, poor CYP2C19 metabolizers had significantly lower sertraline clearance and higher sertraline exposure (as reflected by AUC) than NMs (Wang et al. 2001). Based on these findings, CPIC recommends that, in adults who are CYP2C19 PMs, sertraline and escitalopram doses should be reduced by 50%; however, they caution that extrapolation of their recommendations to pediatric patients be done with increased monitoring (Hicks et al. 2015).
Although some studies in adults with depressive and anxiety disorders suggest relationships between SSRI exposure (or concentrations) (Hiemke et al. 2018) and therapeutic response, only a handful of pediatric studies have examined the relationship between plasma SSRI concentrations and outcome. First, in adolescents with SSRI-resistant major depressive disorder (MDD), higher plasma sertraline and citalopram concentrations were associated with a greater likelihood of response (Sakolsky et al. 2011). Second, in sertraline-treated children and adolescents (N = 90) with MDD or obsessive-compulsive disorder (OCD), there was no relationship between response and plasma sertraline concentrations, although higher plasma concentrations were associated with more antidepressant-related side effects (Taurines et al. 2013). Third, the incidence of adverse events (e.g., activation) may be associated with plasma SSRI concentrations in fluvoxamine-treated youth with generalized, separation, and social anxiety disorders (Reinblatt et al. 2009). Taken together, these findings suggest that plasma SSRI levels are related to side effects and efficacy. Importantly, for sertraline and escitalopram/citalopram, variability in CYP2C19 metabolizer status subtends the relationship between SSRI dose and plasma concentrations.
These findings concerning SSRI dose, plasma concentrations, treatment response, and tolerability relate to a critical question facing clinicians: “Can refining current antidepressant dosing strategies increase treatment response and reduce side effect burden?” In this regard, accumulating data raise the possibility that CYP2C19-guided dosing could decrease SSRI-related adverse events, potentially hasten time to response, and increase the magnitude of response in youth with depressive and anxiety disorders.
Commonly, clinicians initiate SSRIs at low doses and slowly titrate them until either encountering a side effect or response. If intolerable side effects occur, the SSRI dose is decreased or the medication is discontinued. However, although slow titration minimizes side effects, it risks undertreatment in many patients at the initiation of treatment and may ultimately lead to a medication change due to the lack of perceived treatment response. The next SSRI is then chosen through trial and error, based on the clinician's previous experiences or preferences as well as the disorder-specific evidence base for that particular medication (Tulisiak et al. 2017).
The common treatment approaches already described employ a “one size fits all” dosing strategy (i.e., a standard initial dose and a standard dose range) based on average responses in clinical trials. Ultimately, a strategy that tailors dosing based on individual metabolism may accelerate response while decreasing side effects—an immediate benefit to patients and their families. However, there is a dearth of modeling data for PGx-guided SSRI dosing in pediatric patients. With this in mind, we used previously reported 2C19 phenotypes for escitalopram and sertraline to model SSRI exposure (AUC 24) and maximum concentration, C max, across phenotypes, and to develop CYP2C19-guided dosing strategies as an alternative to the current approach: “standard” initial SSRI doses followed by dose titration until either encountering response or treatment-limiting side effects.
Methods
PK modeling and antidepressant dosing simulation
Pediatric volumes of distribution, clearance, and bioavailability were extracted from available data for sertraline and escitalopram. Then, based on sertraline and escitalopram dosing strategies employed in the pediatric registration trials for MDD and OCD, plasma SSRI concentrations were modeled with allometric scaling using MwPharm (MediWare BV, version 3.82) as
PK parameters for a 14-year-old girl with each CYP2C19 metabolizer phenotype were estimated with Bayesian estimation using MW/Pharm (version 3.82, Mediware, Czech Republic) followed by similar modeling in adolescents who were NMs and were aged 12, 13, 15, 16, and 17 years. A published one-compartment PK model developed from healthy volunteers was used as the Bayesian prior for escitalopram (Søgaard et al. 2005) and sertraline (Wang et al. 2001; Rudberg et al. 2008) in combination with allometrically scaled body weight to account for differences in body size. For escitalopram models, parameters included metabolic clearance 33 L/(h · 70kg), V1 17.5 L/kg lean body mass, and ka = 0.8 hours−1. Pediatric PK data from the FDA registration of escitalopram was fitted with the model to ensure accuracy. For sertraline models, parameters included clearance 200 L/(h · 70kg), V1 110 L/kg lean body mass, and ka = 0.8 hours−1. The AUCs were estimated for the final 24 hours during steady state (AUC 24).
Determination of dose ratios based on genotype
CYP2C19-related differences in PK parameters (e.g., AUC 24, C max) for escitalopram and sertraline were determined from the literature and for each phenotype. For poor, intermediate, rapid, and UMs, these parameters were expressed as a ratio of the values in NMs. Then, standard escitalopram and sertraline doses (based on clinical practice, registration trials, or average doses in clinical trials) were evaluated as a ratio of the NM dose for PMs, IMs, RMs, and UMs. In addition, PK models were created for each CYP2C19 phenotype so that phenotype-related differences in AUC 24, C max, and trough concentrations could be explored. These models were based on the sertraline and escitalopram titration schedules from the Pediatric OCD Treatment Study (POTS) and the registration trial for escitalopram in adolescents (aged 12–17 years) with MDD, respectively (Emslie et al. 2009; POTS Team 2004).
Examination of dosing and dosing schedule
For escitalopram, the standard dosing regimen was 10 mg/day for the first 4 weeks, then titration to 20 mg/day for 4 weeks. The pharmacogenetically guided escitalopram regimen was as follows: In PMs, escitalopram was initiated at 5 mg daily and increased to 10 mg daily at week 4. In all other metabolizer groups, escitalopram was initiated at 10 mg daily. IMs were titrated to 15 mg daily at week 4. In NMs, escitalopram was titrated to 20 mg daily at week 4. In RMs, escitalopram was titrated to 15 mg at week 1, and 20 mg at week 2, and 25 mg at week 4. Finally, in UMs, escitalopram was titrated to 15 mg daily at week 1, 20 mg at week 2, 25 mg at week 3, and 30 mg daily at week 4.
The impact of b.i.d. escitalopram dosing was explored as follows: For the RM model, escitalopram was initiated at 10 mg daily × 1 week and then titrated to 15 mg daily × 1 week, 20 mg daily for 1 week, then 10 mg b.i.d. for 1 week, after which it was titrated to 15 mg b.i.d. In the UM model, escitalopram was initiated at 10 mg daily × 1 week and then titrated to 15 mg daily × 1 week, 20 mg daily × 1 week, then 10 mg b.i.d. for 1 week, and titrated to 15 mg b.i.d. for 1 week and then 20 mg b.i.d. For the NM model, escitalopram was initiated at 10 mg daily for 4 weeks and then 20 mg daily for 4 weeks.
For sertraline, the standard dosing regimen consisted of initiation at 50 mg daily, titration to 100 mg/day at week 1, 150 mg/day at week 2, and 200 mg/day at week 3. The PGx-guided sertraline regimen was as follows: in PMs, sertraline was initiated at 25 mg daily, titrated to 50 mg daily at week 1, 75 mg daily at week 2, and 100 mg daily at week 3. In IMs, sertraline was initiated at 25 mg daily, increased to 50 mg daily at week 1, 100 mg daily at week 2, and 125 mg daily at week 3. NMs were initiated at 50 mg daily, increased to 75 mg daily at week 1, 100 mg daily at week 2, and 150 mg daily at week 3. RMs and UMs were initiated at 50 mg, titrated to 100 mg at week 1, 150 mg at week 2, and 200 mg at week 3.
The impact of b.i.d. sertraline dosing was explored in RMs and UMs by modeling. Sertraline was initiated at 50 mg daily × 1 week and then titrated to 100 mg daily for 1 week, after which it was titrated to 100 mg twice daily for 1 week and then 150 mg b.i.d. for the subsequent week to achieve C max and AUC24 that were within 10% of the NMs receiving 200 mg/day. For the NM model, sertraline was initiated at 50 mg daily for 1 week and then 100 mg daily for 1 week and then 150 mg daily for 1 week followed by 200 mg daily for the remainder of treatment.
Results
Antidepressant exposure ratios and target doses
Modeling the standard titration in the different CYP2C19 metabolizer groups predicted the AUC 24 and C max to be higher in PMs and lower in UMs than in NMs (Figs. 1A and 2A). CPIC-defined CYP2C19 phenotype (Caudle et al. 2017) exposure ratios and target doses necessary to achieve an exposure ratio of 1 for poor intermediate RMs and UMs are shown in Table 1 for sertraline and escitalopram. In addition, for UMs and RMs, b.i.d. dosing of escitalopram may achieve AUC 24 and C max that approximate those observed at a 20 mg daily dose in NMs.
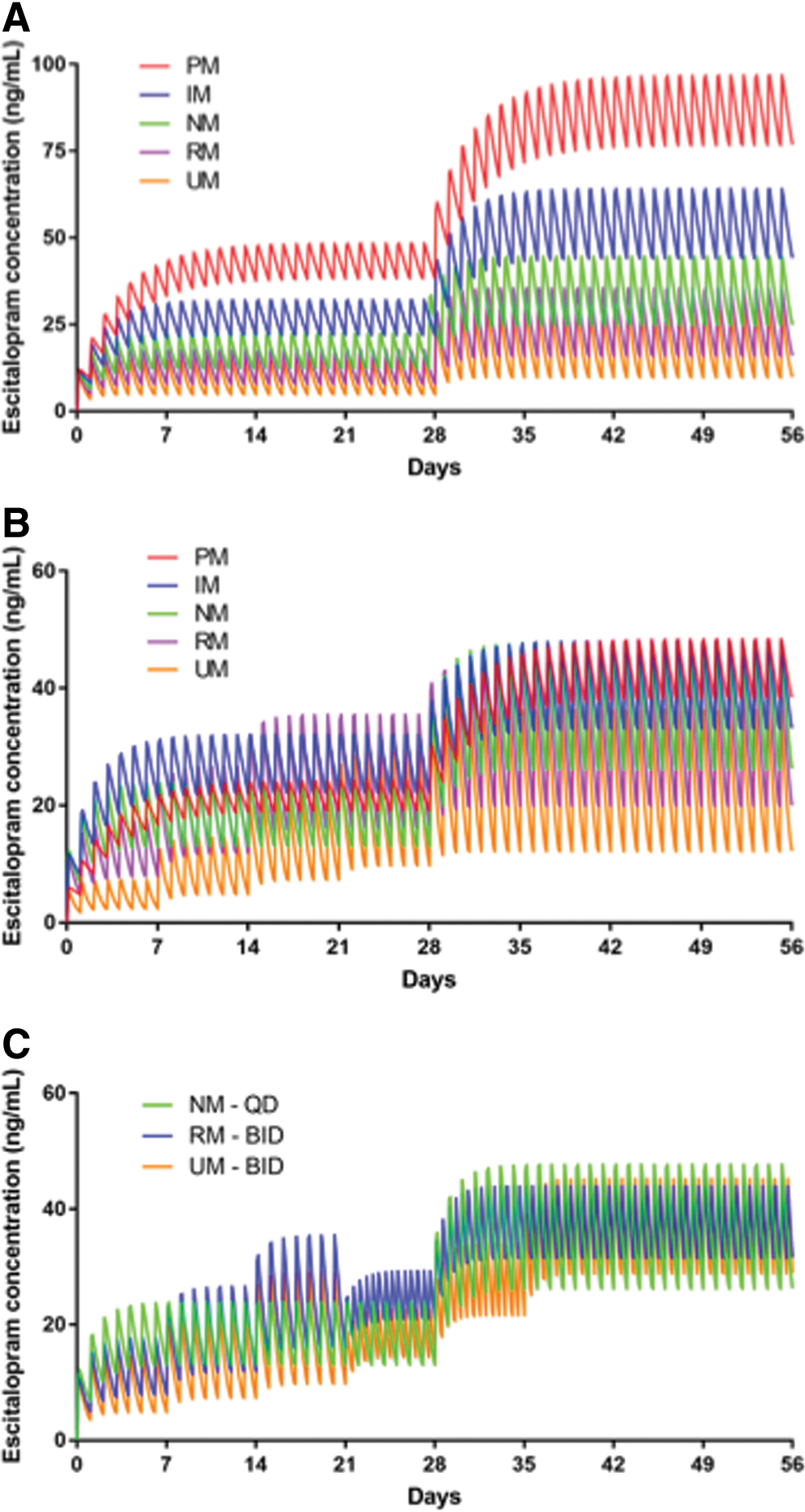
Simulated time course of escitalopram plasma concentrations in adolescents. In the standard dosing model
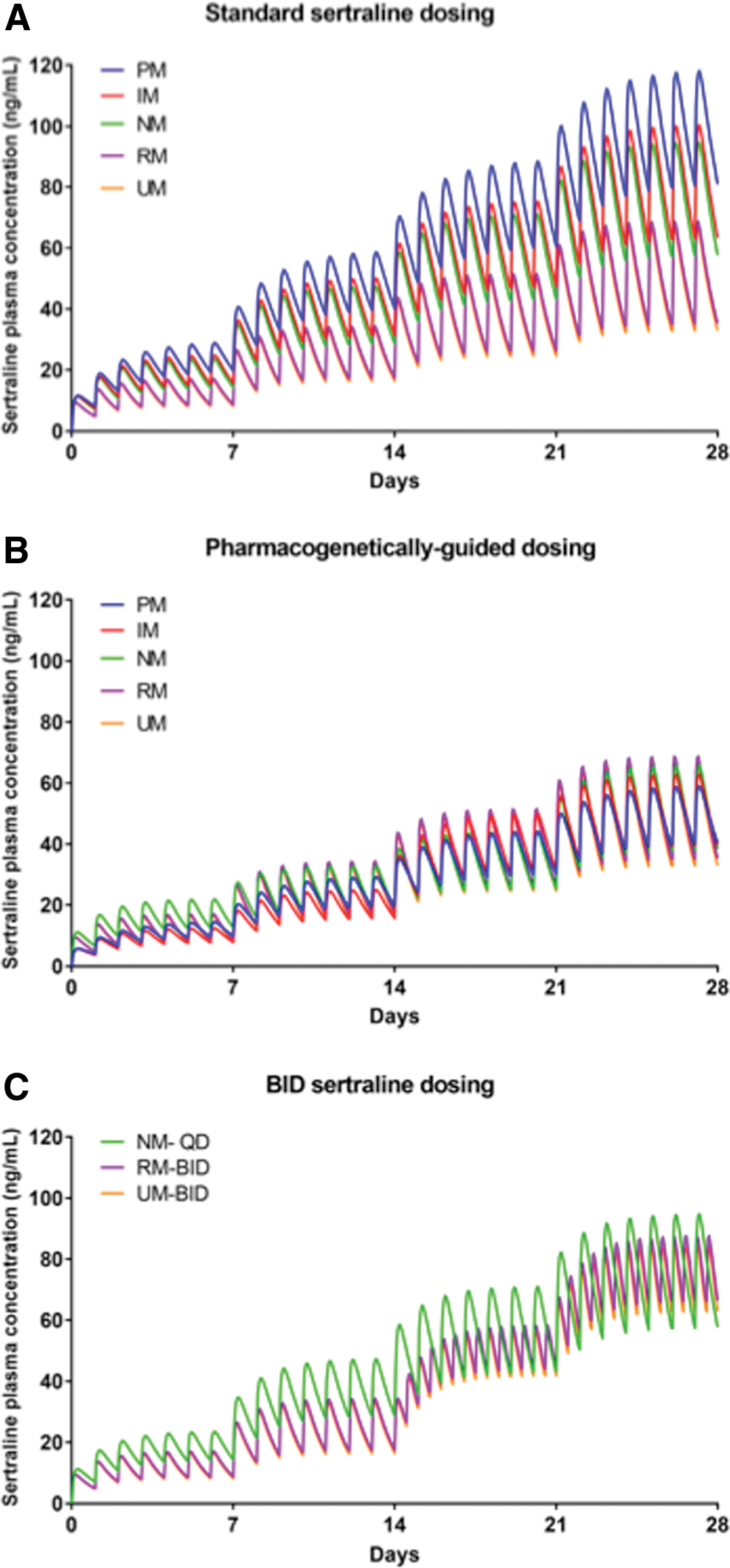
Simulated time course of sertraline plasma concentrations in adolescents. In the standard dosing model
CYP2C19 Phenotype-Determined Exposure Ratios and Selective Serotonin Reuptake Inhibitor Dose Equivalents
Reference doses of 20 and 150 mg/day were used for escitalopram and sertraline, respectively, as these represent the maximum doses in the registration study of escitalopram in adolescent MDD and the approximate average dose in POTS and the Child/Adolescent Anxiety Multimodal Study (CAMS).
Adapted from Chang et al. 2014.
Adapted from Wang et al. 2001.
MDD, major depressive disorder.
To achieve a similar exposure to NMs who began escitalopram at 10 mg daily and increased to 20 mg daily after 4 weeks, CYP2C19-informed dose adjustments were required (Tables 1 and 2, Fig. 1B). For sertraline, to achieve a similar exposure to NMs who began sertraline at 50 mg daily and increased by 50 mg per week to a target dose of 150 mg daily, significant dose adjustments were required (Tables 1 and 3, Fig. 2B).
Pharmacokinetic Parameters in Escitalopram-Treated Adolescents
Cl-m, clearance; t ½, half life; AUC 24, area under the curve (24-hour); q.d., quaque die (once daily); b.i.d., bis in die (twice daily); PGx, pharmacogenetic.
Pharmacokinetic Parameters in Sertraline-Treated Adolescents
Cl-m, clearance; t ½, half life; AUC 24, area under the curve (24-hour); q.d., quaque die (once daily); b.i.d., bis in die (twice daily); PGx, pharmacogenetic.
PK model for escitalopram
Compared with NMs, which had a t ½ of 23.02 hours, the modeled t ½ of escitalopram varied considerably across CYP2C19 phenotypes (PMs: 57.55 hours, IMs 35.97 hours, RMs 16.93 hours, UMs 12.51 hours). Similar relationships between metabolizer phenotype and clearance were also observed (Table 2). For escitalopram-treated adolescents receiving 20 mg/day, C max was higher in PMs (96.78 ng/mL, 217% of NM C max) and in IMs (64.03 ng/mL, 144% of NM Cmax ) than in NMs (44.51 ng/mL), whereas C max was significantly lower in RMs (35.43 ng/mL, 80% of NM C max) and UMs (28.93 ng/mL, 65% of NM C max) (Fig. 1A). Similar phenotype-related relationships were observed for AUC 24 and for C max at the other doses examined (Table 2).
CYP2C19 phenotype-informed escitalopram dosing resulted in similar C max across phenotypes and in similar C max in all groups (48.39 ng/mL in PMs, 48.02 ng/mL in IMs, 44.51 ng/mL in NMs, 44.29 ng/mL in RMs, and 43.4 ng/mL in UMs) (Fig. 1B). For extreme phenotypes, the CYP2C19-guided dosing resulted in C max that were within, respectively, 2.5% and 8% of the C max observed in adolescents who were NMs.
PK model for sertraline
Compared with NMs, which had a t ½ of 22.13 hours, the modeled t ½ of sertraline varied considerably across CYP2C19 phenotypes (PMs: 31.84 hours, IMs 26.08 hours, RMs 20.15 hours, and UMs 19.22 hours). Similar relationships between metabolizer phenotype and clearance were also observed (Table 3). For sertraline-treated adolescents receiving 150 mg daily, C max was higher in PMs (88.35 ng/mL) and IMs (75.26 ng/mL) compared with NMs (66.00 ng/mL), while C max was lower in RMs (51.54 ng/mL) and UMs (49.76 ng/mL) and similar relationships were observed for AUC 24 (Fig. 2A).
For sertraline-treated adolescents receiving 150 mg/day, C max and AUC 24 for PMs were 134% and 143% of those observed in NMs. By contrast, in RMs and UMs, C max and AUC 24 were lower than in NMs (for RMs 78% and 74%, respectively, and for UMs 75% and 71%, respectively).
Finally, the model that leveraged CYP2C19 activity to inform dosing (Figs. 1B and 2B) resulted in similar C max across phenotypes (Tables 2 and 3). For ultrarapid and RMs, b.i.d. dosing of 100 mg sertraline resulted in an AUC 24 that was similar to that observed in NMs receiving 150 mg/day and reduced the C max compared with a 200 mg q.d. dose in the RMs and UMs (Fig. 2C and Table 3). For escitalopram, 15 mg b.i.d. in rapid and 20 mg b.i.d. in UMs resulted in an AUC 24 that was similar to that observed in NMs receiving 20 mg q.d. and reduced the C max compared with a 25 mg q.d. or 30 mg q.d. dose in the RMs and UMs (Fig. 1C and Table 2).
Discussion
This is one of only several studies to pharmacokinetically model antidepressant exposure in youth and the first to evaluate PGx-derived phenotypes on these models. We observed that slower CYP2C19 metabolizers had greater C max and exposure to both sertraline and escitalopram. Furthermore, our models suggest that CYP2C19-informed dosing could normalize sertraline and escitalopram exposure in adolescents. These findings raise the possibility that minimizing CYP2C19-related variability in exposure and clearance might decrease concentration-dependent adverse effects in adolescents who require treatment with sertraline or escitalopram.
With regard to the parameter estimates described herein, our results are generally consistent with the extant steady state PK data in adolescents and shed light on some of the variability of the experimentally determined PK parameters in prior studies. For sertraline, Axelson and colleagues (2002) reported shorter steady state t ½ for sertraline in adolescents treated with 150 mg/day than we described; however, there was considerable variability in the age and body weight of these patients and the patients were treated with between 100 and 150 mg/day (rather than 150 mg/day as in our model). In addition, Axelson and colleagues noted that the t ½ in their study “was substantially shorter than that found in other studies of adolescents.” Interestingly, our results, across CYP2C19 phenotypes in sertraline-treated adolescents, are similar to those reported by Alderman et al. (1998). For escitalopram, our modeled C max and AUC 24 estimates are consistent with the FDA Clinical Pharmacology/Biopharmaceutics Review for Escitalopram (FDA, 2009).
The models described herein structurally differ from many PK models in adults with regard to assumed linear relationships between dose and body weight (Holford and Anderson 2017). In this regard, previous studies suggest that weight-normalized doses (e.g., milligram medication per kilogram body weight) are larger in pediatric patients than in adults (Kearns et al. 2003; Holford and Anderson 2017), and these models predict lower than actual clearance in pediatric patients (Holford and Anderson 2017). Although our models account for weight, like most adult models, they also incorporate allometric scaling, which reflects physiologic characteristics that are affected by development (Kearns et al. 2003). This scaling may account for differences between the CYP2C19-related differences in clearance and exposure in our models and CYP2C19-related parameter estimates in adults (Steere et al. 2015). Regarding the impact of age on clearance and exposure, as the age (and weight) increases, the exposure and C max for both escitalopram and sertraline decrease. The biggest increase in body size occurs between 12 and 14 years of age, and the exposure is ∼14% lower in 12-year-old adolescents than in 14-year-old adolescents (during which time weight—at the 50th percentile—increases by 8 kg). By contrast, the difference in exposure between 14 and 17 years of age is only 8%. The magnitude of these differences is much less than the differences between metabolizer groups (escitalopram exposure is 158% higher in PMs than in NMs, and 48% lower in UMs than in NMs).
The relationship between PK parameters (e.g., t½ ) and tolerability (e.g., suicidality) has received considerable attention since the 2004 black box warning for antidepressants in children, adolescents, and young adults. Using overlapping data sets, several studies observed associations between the relative risk of treatment-emergent suicidality and t ½ (Smith 2009; Rahn et al. 2015). In the first of these studies, antidepressant t ½ and the relative risk of treatment-emergent suicidality significantly correlated (r = 0.786, p = 0.036). However, this study used adult t ½ data secondary to “an unavailability of published information [for pediatric t ½] (Smith 2009).” A second study replicated this relationship between SSRI t 1/2 and treatment-emergent suicidality (Rahn et al. 2015). However, the latter study leveraged a similar PK modeling approach as described herein to simulate plasma SSRI concentrations for paroxetine, citalopram, sertraline, and fluvoxamine. Then, the authors developed an SSRI titration regimen to mirror the PKs of fluoxetine, the SSRI with the least treatment-emergent suicidality, and the longest t ½.
Our CYP2C19-guided model as well as the dosing strategy reported herein extends the work of Rahn et al. (2015) by using a model that directly addresses differences in exposure not just based on medication type and patient population, but also based on CYP2C19-related differences in metabolism. Rahn et al. suggested that antidepressants with longer half-lives (and, therefore, higher dose-related exposure) are associated with a lower risk of adverse events than medications with a short half-life. However, although this recommendation emphasizes the importance of half-life, it may not fully account for differences in dose-related exposure that impact antidepressant tolerability in youth.
The relationship between SSRI exposure and treatment response that have been observed in adolescents with treatment-resistant MDD (Sakolsky et al. 2011) and meta-analyses suggests that SSRI dose is related to the trajectory of response in pediatric patients with anxiety disorders (Strawn et al. 2018). In addition, several lines of evidence suggest a relationship between SSRI dose (or plasma SSRI concentration) and side effects in pediatric patients. For example, in sertraline-treated children and adolescents (N = 90), higher plasma concentrations were associated with more antidepressant-related side effects (Taurines et al. 2013), and activation is associated with plasma fluvoxamine concentrations in anxious youth (Reinblatt et al. 2009). More recently, a retrospective study of escitalopram/citalopram tolerability (N = 248) showed CYP2C19 metabolizer status significantly influenced weight gain, activation, and insomnia (Aldrich et al. 2019), suggesting that side effects were related to CYP2C19-related differences in exposure rather than differences in dosing (despite no observed differences in dosing across phenotypes). Taken together, these findings suggest that plasma SSRI concentrations might be related to side effect burden and potentially efficacy. Thus, for clinicians, differences in CYP2C19 activity may subtend the differences in sertraline and escitalopram/citalopram exposure. As such, CYP2C19-guided dosing for escitalopram and sertraline could refine the “unpredictable” variability in exposures related to side effects and differences in treatment response; however, there will still be variability not explained by CYP2C19. It is further noteworthy that the POTS (sertraline) team 2004) employed what some have considered to be an aggressive dosing strategy, whereas the mean titrated dose of the CAMS study (sertraline) (Walkup et al. 2008) was >130 mg/day. The dosing strategies and mean treatment dose, and ostensibly higher exposure (compared with typical clinical practice), may have both contributed to increased efficacy, but may have also adversely affected tolerability. However, without genotype data from the patients, this is difficult to determine.
Limitations
Although this is the first PK modeling study of SSRIs in children and adolescents to examine CYP2C19-related differences in SSRI metabolism and exposure, several important limitations warrant additional discussion. First, standard assumptions related to model parameters were necessary, and although they can be justified by the central limit theorem and the maximum entropy principle, the availability of the raw data would have allowed testing of these assumptions. Second, model assumptions may limit the generalizability of our findings. In this regard, our models assume 100% adherence and that medications are taken at the same time each day; however, antidepressant adherence in adolescents is frequently less than desired (Woldu et al. 2011). Third, clinical and medication-specific endogeneity among model parameters is difficult to discern. For example, if a patient experiences side effects that are related to C max or a lack of efficacy associated with reduced exposure, it remains likely that adherence be decreased, thus affecting C max and AUC 24. Fourth, additional CYP or non-CYP factors may affect the metabolism of these medications (e.g., CYP2B6, and phenoconversion as a result of concomitant medications) (Klieber et al. 2015). Fifth, our models examined sertraline and escitalopram in adolescents rather than in children and adolescents. Our focus on adolescents is based on the FDA approval for escitalopram in adolescents, and for sertraline, it is based on greater C max and AUC 24 in children than in adolescents, although these age-related differences disappear when model parameters incorporated body weight (Alderman et al., 1998).
Conclusions
Building PK models in pediatric patients that take into account metabolizer status may facilitate optimal dosing regimens that could be used to predict individual SSRI exposure. Modeling approaches such as those leveraged herein may provide a critical scaffold on which improvements in clinical practice and clinical trial design can occur. However, refining these models ultimately requires plasma SSRI concentrations in adolescents with known CYP2C19 phenotypes. Ultimately, moving beyond the “one size fits all” approach to pharmacogenetically-guided dosing may enhance antidepressant treatment response, safety, and tolerability in youth. However, to answer the question, “Can refining current antidepressant dosing strategies increase treatment response and reduce side effect burden?” we require prospective trials that compare pharmacogenetically-guided dosing strategies with standard approaches.
Clinical Significance
The models described herein suggest that for pediatric patients who are CYP2C19 PMs, sertraline and escitalopram might be initiated at lower doses, whereas in youth with increased CYP2C19 activity (e.g., UMs), higher doses may be required. For example, an UM requires an escitalopram dose of 30 mg/day to approximate the exposure seen in an NM treated with 20 mg/day, whereas for sertraline, an UM requires 200 mg/day to approximate the exposure associated with a 150 mg/day dose in an NM. However, while increasing the SSRI dose to maintain adequate exposure in adolescents with increased CYP2C19 metabolism, our models suggest that this would also increase C max. Such increases in C max ostensibly increase side effects associated with peak SSRI levels. Therefore, based on the modeling presented herein, in some individuals with increased metabolism, b.i.d. dosing might be considered to maintain exposure without increasing C max, consistent with prior suggestions of b.i.d. sertraline and escitalopram dosing (Findling et al. 2006). Ultimately, prospective trials of pharmacogenomically guided dosing in the pediatric population are needed to extend the findings of these modeling studies and to examine the safety of doses that exceed the FDA-approved guidelines for this age range.
Footnotes
Disclosures
Dr. Strawn has received research support from Edgemont, Eli Lilly, Shire, Allergan, Lundbeck, and the National Institutes of Health (NIMH and NIEHS) and Neuronetics. He receives royalties from Springer Publishing and has received material support from and provided consultation to Myriad/Assurex. He has received honoraria from CMEology and UpToDate. Dr. Ramsey has received travel support from the American Academy of Child & Adolescent Psychiatry. Mr. Poweleit has no financial conflicts of interest.