
Abstract
Objective:
In the U.S. ∼33% of children with attention-deficit/hyperactivity disorder (ADHD) are diagnosed during their preschool years (<6 years of age). The majority of these children are treated with a psychopharmacological treatment, despite limited data on pharmacokinetics (PKs), efficacy, or safety of these medications in this population. A phase 4, single-dose open-label study was conducted to assess the PK profile of amphetamine extended-release orally disintegrating tablets (AMP XR-ODT) under fasted conditions in preschool-aged children with ADHD.
Methods:
Preschool-aged children (aged 4 to <6 years) with a confirmed ADHD diagnosis were enrolled and administered AMP XR-ODT 3.1 mg under fasted conditions. Plasma samples were analyzed for d- and l-amphetamine (AMP) via liquid chromatography–tandem mass spectrometry. Area under the concentration-time curve from time 0 extrapolated to infinity (AUC0–inf), area under the concentration-time curve from time 0 to the last measurable plasma concentration (AUC0–T), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), terminal half-life (t1/2), apparent volume of distribution (Vz/F), and apparent clearance (CL/F) for d- and l-AMP and safety were assessed.
Results:
The PK and safety analyses included 15 preschool-aged children (4 years old, n = 6; 5 years old, n = 9); 14 completed the study. Quantifiable plasma concentrations for d- and l-AMP were observed 1.5 hours postdose and throughout the 24-hour sampling period. For d- and l-AMP, mean AUC0–inf was 315.2 and 104.4 h·ng/mL, AUC0–T was 296.0 and 96.8 h·ng/mL, t1/2 was 8.0 and 9.2 hours, Cmax was 23.0 and 7.0 ng/mL, Tmax was 3.9 and 4.0 hours, CL/F was 6996.3 and 6837.1 mL/h, and Vz/F was 75,874.5 and 84,140.0 mL, respectively. Adverse events included tachycardia (n = 2), neutropenia (n = 1), increased alanine aminotransferase (n = 1), and aspartate aminotransferase (n = 1).
Conclusions:
AMP XR-ODT 3.1 mg was well tolerated in preschool-aged children, with detectable plasma AMP concentrations over 24 hours, and a PK profile consistent with once-daily dosing.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a common neurobehavioral condition affecting ∼7% of children globally with a median age of diagnosis of 7 years of age (Wolraich et al. 2011; Thomas et al. 2015). Although establishing a definitive ADHD diagnosis in preschool-aged children can be challenging, ∼33% of children with ADHD in the United States are diagnosed during their preschool years (Wigal and Wigal 2007; Visser et al. 2015).
In preschool-aged children with ADHD, early intervention is imperative to minimize continued or future behavioral difficulties that can impair subsequent social and academic performance. The American Academy of Pediatrics and the American Academy of Child and Adolescent Psychiatry recommend behavior therapy as the first-line treatment for ADHD in preschool-aged children (aged 4–5 years) (Pliszka 2007; Wolraich et al. 2011). The use of stimulant medication is only recommended in cases where initial behavior therapy proves unsuccessful following at least 8 weeks of parent and/or teacher management (Gleason et al. 2007; Wolraich et al. 2011). Although pharmacotherapy is used in ∼18% of preschool-aged children with ADHD (Danielson et al. 2018), only immediate-release amphetamine (AMP) formulations are approved by the U.S. Food and Drug Administration (FDA) for the treatment of ADHD in this age group (Childress and Stark 2018). Of preschool-aged children with ADHD who are prescribed medication for ADHD, ∼91% are prescribed ADHD medications that have not been specifically studied in this population (Panther et al. 2017). Consequently, there is a great need to better understand the pharmacokinetic (PK) profile of extended-release formulations in this population, as well as to conduct additional studies supporting safe and effective prescription practices in the preschool-age population.
PK studies in preschool-aged children of medications developed for older children and adults are needed to assess the impact of variations in physiology, such as body water content and organ system development, on the PK profile (Kearns et al. 2003; Wells et al. 2005; Childress and Stark 2018). Accordingly, the FDA issued guidelines in agreement with the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act to ensure age-appropriate studies regarding the use of medications in pediatric populations (Food and Drug Administration 2014, 2016a).
Amphetamine extended-release orally disintegrating tablets (AMP XR-ODT) are approved as a once-daily dose for the treatment of ADHD in children aged 6 years and older (Stark et al. 2017). AMP XR-ODT comprises both d- and l-AMP enantiomers in a 3:1 ratio, and the orally disintegrating tablet (ODT) formulation is based on cation exchange resin technology, which facilitates ∼50% immediate release and 50% delayed release of AMP (Stark et al. 2017). The ODT disintegrates in the mouth, and any remaining fragments can be swallowed with saliva (Stark et al. 2017). Previous studies demonstrated that an 18.8 mg dose of AMP XR-ODT was well tolerated in 6- to 12-year-old children with ADHD and displayed a PK profile consistent with once-daily dosing (Stark et al. 2017). Here, we report the results of a phase 4, single-dose PK and safety study of AMP XR-ODT 3.1 mg in 4- to 5-year-old children, which was conducted in response to a request by the FDA for additional PK data in preschool-aged children of an ADHD medication that is approved for use in children aged >6 years (Food and Drug Administration 2016b).
Methods
Study design
This open-label, multicenter, single-dose PK study was conducted at two U.S. sites in compliance with local regulatory requirements and in accordance with the ethical principles as stated in the Declaration of Helsinki, current Good Clinical Practices, and the U.S. Investigational New Drug regulations (U.S. 21 Code of Federal Regulations, part 312). All protocols, informed parental consent forms, and information provided to parents/legal guardians were approved by each center's institutional review board before study initiation. Before enrollment, written informed consent was obtained from the parent or legal guardian of each patient. Participants could withdraw from the study at any time, for any reason.
Before dosing, participants were screened for eligibility. Clinical laboratory testing, including urinalysis for the presence of AMPs and methylphenidate; vital signs evaluation; 12-lead electrocardiogram (ECG); and physical examination were conducted. The Kiddie Schedule for Affective Disorders and Schizophrenia–Present and Lifetime version (K-SADS-PL), a semi-structured interview, was administered to confirm a diagnosis of ADHD and to determine whether psychiatric comorbidities were present. Additionally, a suicidality assessment, via a semi-structured interview, was completed with the parent or guardian. Participants were required to discontinue stimulant use at least 5 days before dosing; the use of nonstimulant medication required a washout period of at least five half-lives, except for fluoxetine, for which a 28-day washout period was required.
Following a 10-hour overnight fast at the study center, all eligible participants were administered a single dose of AMP XR-ODT 3.1 mg. Participants were required to refrain from drinking water for at least 1 hour before and 1 hour after dosing. In addition, participants could not eat for 3 hours following administration. A mouth rinse with 60 mL of water was permitted before study drug administration. Participants were instructed to move the tablet around their mouth to facilitate disintegration without crushing or chewing and then swallow any remaining fragments with saliva. A mouth check was performed to ensure that the ODT had disintegrated and all fragments had been swallowed. Serial blood samples were collected in-house through 12 hours postdose for the assessment of plasma concentrations of d- and l-AMP. Participants were permitted to go home after the 12-hour PK sample was drawn. The final PK sample collection and end-of-study assessments were conducted on day 2 at 24 hours postdose, with an 18- to 30-hour window permitted.
Inclusion/exclusion criteria
Eligible participants were aged 4 to <6 years at the time of screening, had both parents or legal guardians who could speak English and could provide written parental consent, and met the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria for ADHD, combined, hyperactive/impulsive, or inattentive profile (confirmed using the K-SADS-PL) (American Psychiatric Association 2013). If the ADHD diagnosis was difficult to establish, the participant was not enrolled. Other inclusion criteria included the absence of clinically significant abnormalities that would preclude treatment with AMP XR-ODT, not taking any ADHD medication or had a clinical indication for withdrawing from their current ADHD medication, and could refrain from engaging in strenuous exercise during the study.
Exclusion criteria included a history of seizure disorder; suicidal ideation or behavior; violent or disruptive behavior; clinically significant medical conditions, including cardiovascular or psychiatric disorders; history of anxiety or aggression; hepatitis B or C; HIV; evidence of physical, sexual, or emotional abuse; history of parental bipolar disorder; living with parent/caregiver currently abusing stimulants or cocaine, or with a history of substance abuse within the last 12 months; unable to ingest AMP XR-ODT; clinically significant laboratory results at screening or before dosing; clinically significant ECG results at screening; height or weight ≤5th percentile for age or sex at screening; blood pressure (BP) ≥95th percentile for age, sex, and height at screening or before dosing; a history of Tourette syndrome or tics; received pharmaceutical drugs, including monoamine oxidase inhibitors, coumarin anticoagulants, tricyclic antidepressants, selective serotonin inhibitors, herbal remedies, pain medication (except nonsteroidal anti-inflammatory drugs or acetaminophen), antihistamines, and proton-pump inhibitors or antacids; a documented allergy or hypersensitivity to AMP; history of adverse reactions to stimulant treatments; medical condition that would preclude the use of a stimulant drug; or had participated in an investigational drug study within the past 6 months.
Medication
AMP XR-ODT was provided as a single tablet, containing 3.1 mg of AMP base (with d- and l-AMP in a 3:1 ratio). The starting dose in pediatric patients aged 6–17 years is 6.3 mg (Adzenys XR-ODT 2018). The lowest dose (3.1 mg) was selected for use in this single-dose PK study in children aged 4 to <6 years. All medications were administered at each of the study sites under supervision of the site staff to ensure compliance.
During the study, the use of asthma inhalers, including corticosteroid and rescue bronchodilator preparations, was permitted. However, participants were not allowed to use the over-the-counter medications and nutritional supplements for at least 7 days before study initiation through the end of the study. Furthermore, consumption of beverages, foods, or medications containing alcohol, orange juice, fruit juice, or caffeine was prohibited 48 hours before dosing until the end of the study.
Sample collection and PK analysis
In total, 10 blood samples were collected for PK analysis from each participant: 0 (predose), 1.5, 3, 4, 6, 7, 8, 10, 12, and 24 hours postdose. On day 1, 9 samples were collected; the final 24-hour sample was collected on day 2 with sample collection permitted between 18 and 30 hours postdose. Plasma samples were analyzed for d- and l-AMP using a validated liquid chromatography–tandem mass spectrometry assay, as previously reported (Stark et al. 2017).
Plasma PK parameters for d- and l-AMP included area under the concentration-time curve from time 0 extrapolated to infinity (AUC0–inf); area under the concentration-time curve from time 0 to the last measurable plasma concentration (AUC0–T); maximum plasma concentration (Cmax); time to maximum plasma concentration (Tmax); apparent clearance (CL/F); terminal half-life (t1/2); and apparent volume of distribution (Vz/F). PK parameter calculations were performed using individual plasma concentration–time data, and concentrations below the limit of quantification (BLQ) occurring before Tmax were set to zero. However, if the BLQ occurred between two measurable concentrations, or after Tmax, it was set to missing, consistent with previous report (Stark et al. 2017).
Safety analysis
Safety and tolerability assessments included vital signs evaluation, 12-lead ECG, physical examination, clinical laboratory tests, assessment of suicidality, and observed or reported adverse events (AEs). Patients were monitored for AEs from the time of informed consent through the end of the study. All reported or documented AEs were treated and followed until resolution. Any AEs occurring before administration of study medication were included in the medical history; events that occurred following drug administration were captured as treatment-emergent AEs (TEAEs).
Statistical analyses
All analyses were conducted using SAS (release 9.4), and PK parameter estimations were performed using Phoenix WinNonlin® software (version 8.0; Pharsight, Cary, NC). Safety data were summarized using descriptive analysis. To achieve a 95% confidence interval within 60% and 140% of the geometric mean estimates for CL/F and Vz/F with a minimum of 80% power for the both d- and l-AMP, a sample size of five or fewer participants was required. To account for participants who may terminate early or were unevaluable, a minimum of six to eight participants was needed to complete the study.
Results
Participants
Of 22 participants screened, 15 received AMP XR-ODT 3.1 mg and were included in the PK and safety analyses. A total of 14 participants completed the study; one participant did not return on day 2 for the 24-hour blood draw and was lost to follow-up. Demographic and baseline characteristics are shown in Table 1. The majority of participants were 5 years of age (9/15, 60%), male (9/15, 60%), and black/African American (13/15, 86.7%). The median body mass index of the participants was 16.6 kg/m2 (range 14.0–19.8 kg/m2).
Demographic and Baseline Characteristics
AMP XR-ODT, amphetamine extended-release orally disintegrating tablet; BMI, body mass index; n, number of participants; SD, standard deviation.
PK assessments
Quantifiable plasma concentrations of both d- and l-AMP were observed at the first postadministration time point (1.5 hours) and throughout the 24-hour sampling period (Fig. 1). Maximum plasma concentrations were observed at 4 hours after AMP XR-ODT administration.
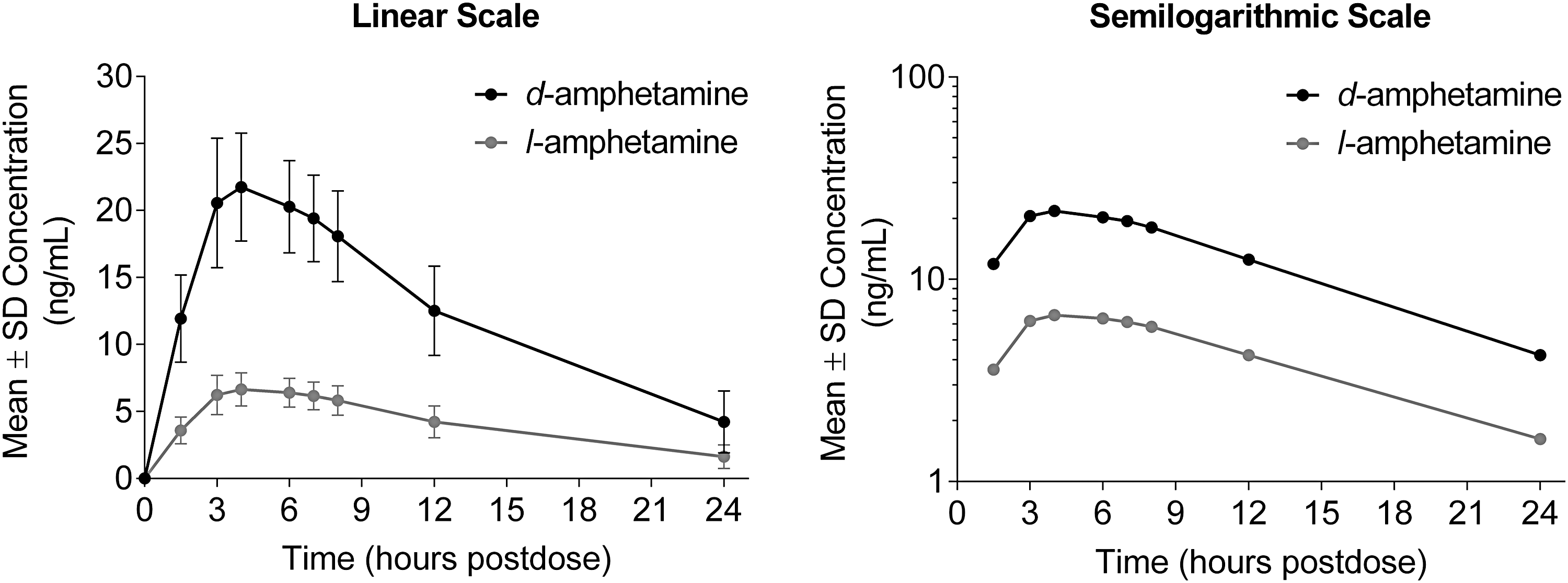
Mean plasma concentration–time profiles for d- and l-amphetamine after administration of AMP XR-ODT under fasted conditions in preschool-aged children with ADHD. ADHD, attention-deficit/hyperactivity disorder; AMP XR-ODT, amphetamine extended-release orally disintegrating tablet; SD, standard deviation.
PK parameters for d- and l-AMP are presented in Table 2. The mean total exposure (AUC0–inf), overall exposure (AUC0–T), and Cmax were higher for d-AMP compared with l-AMP after oral administration, as expected. Other parameters were comparable.
Pharmacokinetic Parameters for d- and l-Amphetamine
AMP XR-ODT, amphetamine extended-release orally disintegrating tablet; AUC0–inf, area under the concentration-time curve from time 0 extrapolated to infinity; AUC0–T, area under the concentration-time curve from time 0 to the last measurable plasma concentration; CL/F, apparent clearance; Cmax, maximum plasma concentration; max, maximum; min, minimum; PK, pharmacokinetic; SD, standard deviation; t1/2, terminal half-life; Tmax, time to maximum plasma concentration; Vz/F, apparent volume of distribution.
Safety assessments
Patient AEs are summarized in Table 3. Two participants experienced ≥1 TEAE (Table 3). There were no serious AEs, AEs leading to discontinuation, or deaths. All AEs were mild and resolved. TEAEs included tachycardia (n = 2), alanine aminotransferase (ALT) increase (n = 1), aspartate aminotransferase (AST) increase (n = 1), and neutropenia (n = 1). Tachycardia was considered nonserious, mild in severity, and resolved. ALT and AST levels in one subject were normal at screening, increased postadministration (day 1), and remained out-of-range 24 hours postdose on day 2, but returned to within the normal range when tested a second time on the same end-of-study day (day 2).
Treatment-Emergent Adverse Events
Normal values were recorded at the second testing performed at the end of the same study day.
ALT, alanine aminotransferase; AMP XR-ODT, amphetamine extended-release orally disintegrating tablet; AST, aspartate aminotransferase; TEAE, treatment-emergent adverse event.
Serum chemistry parameters remained within the normal range from baseline to day 2 for all participants, except for ALT and AST, as noted above. Systolic BP, diastolic BP, and heart rate increased slightly from baseline beginning at 1 hour postdose, peaking at 4 hours postdose, and declining at 8 hours postdose on day 1; however, levels returned to baseline on day 2. ECG results were normal in most participants; all abnormal findings were not considered clinically significant. Physical examinations and remaining clinical laboratory analyses revealed no clinically significant findings. No suicidal ideation or behavior was reported before or after administration of AMP XR-ODT.
Discussion
This study was conducted as a postmarketing requirement for AMP XR-ODT to address the need for additional PK data in children aged 4–5 years and to satisfy the Pediatric Research Equity Act requirements. To the best of our knowledge, this is the first PK study of an extended-release orally disintegrating AMP in preschool-aged children (aged 4 to <6 years) under fasted conditions. The PK profile in the preschool-aged children of AMP XR-ODT was similar to those previously reported in children aged 6–7 years (Stark et al. 2017). Peak plasma concentrations for d- and l-AMP were achieved within a few hours after administration, followed by a slow decline over 24 hours, indicative of an extended-release formulation (Stark et al. 2017). In the preschool-age group, the elimination half-life was 1–2 hours shorter than that previously reported for 6- to 12-year-old children administered 18.8 mg AMP XR-ODT (d-AMP: 8.0 vs. 9.5 hours; l-AMP: 9.2 vs. 11.0 hours) (Stark et al. 2017). Overall, the data indicate that there are no major differences in the AMP PK profile or safety of 4- to 5-year-old children compared with school-aged children after administration of AMP XR-ODT. Consistent with previous findings in the 6- to 12-year-old age group (Stark et al. 2017), AMP XR-ODT was generally well tolerated in the preschool-age group.
To the best of our knowledge, there are no published PK studies of AMP medications in preschool-aged children. Most AMP formulations contain both the d- and l-isomers (Markowitz and Patrick 2017); d-AMP is three- to five-fold more potent in releasing dopamine than l-AMP, which is more potent in releasing noradrenaline, and both isomers contribute to the effectiveness of AMP in the treatment of ADHD (Heal et al. 2013). A 3:1 ratio of d- and l-AMP is thought to optimize the entry of the d-isomer into catecholaminergic nerve terminals, due to competition for the dopamine transporter by the l-isomer, causing prolonged d-AMP-mediated neurotransmitter release (Heal et al. 2013). Differences in the disposition of the AMP isomers, as reflected in the elimination half-life, indicate that the t1/2 of d-AMP is shorter than that for l-AMP, which is consistent with the PKs of AMP in other formulations (Markowitz and Patrick 2017). Most AMP PK studies have shown that body weight greatly influences AMP plasma concentrations in various age groups (Markowitz and Patrick 2017), although weight was not reported to be a major factor in AMP exposure following AMP XR-ODT administration in 6- to 12-year-old children (Stark et al. 2017). The preschool-aged children in the current study received six times less AMP XR-ODT than the 6- to 12-year-old children (3.1 vs. 18.8 mg) (Stark et al. 2017); therefore, a direct comparison of systemic exposure cannot be made (Markowitz and Patrick 2017).
Currently, three immediate-release formulations of AMP are approved by the FDA for use in children aged 3–5 years with ADHD (Adderall 2016; Evekeo 2016; Procentra 2017). However, efficacy and safety trials supporting their use in this age group are lacking (Childress and Stark 2018). Due to the absence of data on the PK profile and safety of AMP medications in preschoolers, this study was conducted with the lowest available dose of AMP XR-ODT. In contrast, a dose of 18.8 mg AMP XR-ODT was previously used to assess the PK profile in 6- to 12-year-old children (Stark et al. 2017).
Conclusions
This study provides important information regarding PK parameters of AMP XR-ODT in the preschool-age population. AMP XR-ODT exhibited a PK profile in children aged 4 to <6 years consistent with once-daily dosing and was well tolerated with no major safety concerns.
Clinical Significance
Despite a lack of age-appropriate information, children aged 4 to <6 years with ADHD are treated with ADHD medications; therefore, it is important to provide data regarding safety, efficacy, and PK in this age group to facilitate age-appropriate clinical prescribing decisions. AMP XR-ODT, a novel orally disintegrating formulation of AMP, is approved for children aged 6–12 years and adults. This was a preliminary PK study demonstrating that AMP XR-ODT provides a PK profile consistent with once-daily dosing and is generally well tolerated in the 4- to <6-year-old age group. These findings support additional efficacy and safety studies of AMP XR-ODT in preschool-aged children with ADHD.
Footnotes
Acknowledgments
The authors thank the study participants as well as Dr. Robert Riesenberg, MD (Atlanta Center for Medical Research, Atlanta, GA), for their invaluable contribution to this study. Medical writing and editorial support were provided by Marie-Louise Ricketts, PhD, and Nicole Seneca, PhD, of AlphaBioCom, LLC (King of Prussia, PA).
Disclosures
A.M., T.L., A.V., and A.H. have nothing to disclose. D.E. was an employee of Neos Therapeutics, Inc., at the time this study was conducted and currently holds stock in Neos Therapeutics, Inc. C.R.S. is an employee of Neos Therapeutics, Inc., with stock options in Neos Therapeutics, Inc., and stock in Pfizer.