
Abstract
Objective:
To examine the efficacy, safety, and tolerability of methylphenidate extended-release orally disintegrating tablets (MPH XR-ODT) for the treatment of attention-deficit/hyperactivity disorder (ADHD) during the open-label dose-optimization/stabilization period of a phase 3 laboratory classroom study.
Methods:
Children (6–12 years) diagnosed with ADHD were enrolled. Treatment was initiated with MPH XR-ODT 20 mg daily. Doses were adjusted weekly by 10–20 mg during the 4-week dose-optimization period (visits 2–5) until an optimal dose was reached. The optimal dose was sustained during a 1-week stabilization period (visits 6–7). Efficacy was assessed using the ADHD Rating Scale-IV (ADHD-RS-IV) score and the Clinical Global Impression-Improvement (CGI-I) score. Adverse events (AEs) were recorded throughout the study. A secondary subgroup analysis by baseline ADHD-RS-IV score, sex, age, and weight was also performed.
Results:
The mean (standard deviation [SD]) final optimized MPH XR-ODT daily dose was 41.8 (14.6) mg and ranged from 20 to 60 mg. Final optimized dose was higher for children with more severe baseline ADHD-RS-IV total scores. ADHD-RS-IV total scores decreased progressively during dose optimization, with a mean (SD) change from baseline at visit 7 of −21.4 (8.9). CGI-I scores shifted from “minimally improved” (mean [SD]: 3.1 [1.1]) at visit 3 to “much improved” (1.6 [0.6]) at visit 7. Baseline ADHD-RS-IV total score was highest for participants optimized to 40 mg (mean [standard error]: 40.0 [1.4]) and lowest for those optimized to 20 mg (34.8 [2.1]). By visit 6, mean ADHD-RS-IV score was comparable for all optimized dose groups. Common treatment-emergent AEs (≥5% of participants) included decreased appetite, upper abdominal pain, headaches, and insomnia.
Conclusions:
Dose optimization of MPH XR-ODT led to a reduction in ADHD symptoms, indicated by a decrease in ADHD-RS-IV and CGI-I scores. AEs were consistent with those of other MPH products.
Clinical Trial Registry: NCT01835548 (ClinicalTrials.gov).
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is one of the most common neurobehavioral disorders affecting children globally (Albert et al. 2017; Raman et al. 2018) and is characterized by inattention, hyperactivity, and impulsivity (Holbrook et al. 2016). Childhood ADHD can negatively impact quality of life and affect outcomes in education and work (Peasgood et al. 2016).
Both the American Academy of Pediatrics and the American Academy of Child and Adolescent Psychiatry recommend psychopharmacological therapy with United States Food and Drug Administration (FDA)-approved medication for ADHD and/or behavior therapy for children (6–11 years of age) and adolescents (12–18 years of age) (Pliszka and The AACAP Work Group on Quality Issues 2007; Wolraich et al. 2019). Psychopharmacological therapy can help ADHD symptoms, resulting in better classroom performance and quality of life in children with ADHD (Barbaresi et al. 2007; Childress et al. 2017). Stimulants are recommended as first-line pharmacological treatment due to extensive evidence of efficacy (Pliszka and The AACAP Work Group on Quality Issues 2007; Wolraich et al. 2019). Treatment typically starts with a recommended dose, followed by dose titration to achieve an optimal response in symptom reduction, while balancing adverse events (AEs) (Pliszka and The AACAP Work Group on Quality Issues 2007; Wolraich et al. 2019).
A once-daily, extended-release orally disintegrating tablet (XR-ODT) formulation of methylphenidate (MPH) is FDA approved for ADHD treatment in children 6–17 years of age (Neos Therapeutics, Inc. 2017). The XR-ODT formulation uses ion-exchange resin technology to create stable MPH microparticles; the positively charged MPH molecule replaces the positively charged mobile ion of the exchange resin (Na+) when the MPH salt is dissolved. Coated (extended release) and uncoated (immediate release) MPH microparticles are then compressed into ODTs. In a randomized, double-blind laboratory classroom study, MPH XR-ODT (at doses of 20–60 mg) significantly improved ADHD symptoms vs placebo, with an early onset of efficacy and long duration of action (Childress et al. 2017). After screening, MPH XR-ODT was started at a 20-mg dose and could be increased in 10- to 20-mg increments over a 4-week open-label period until an optimal dose, based on efficacy and tolerability, was achieved or a maximum dose of 60 mg per day was reached. The optimized dose was maintained for a 1-week stabilization period, and participants were then randomized (1:1) to either the optimized dose of MPH XR-ODT or placebo (Childress et al. 2017).
Although a dose-optimization period is common in ADHD laboratory classroom studies, few studies describe the efficacy and safety outcomes of the dose-optimization period (Childress et al. 2015, 2017; Huss et al. 2017; Wigal et al. 2018). Laboratory classroom studies with dose-optimization periods have shown that only a small percentage of participants remain on the initial dose (Murray et al. 2011; Childress et al. 2017; Wigal et al. 2018). Furthermore, while guidance on timing of dose adjustments for dose optimization is provided in the label for most approved ADHD medications, the time to a stable dose varies widely in clinical practice (Janssen Pharmaceuticals, Inc. 2017; Neos Therapeutics, Inc. 2017; Rhodes Pharmaceuticals L.P. 2019; Pasadyn et al. 2020). Understanding patterns of symptom improvement during dose optimization could guide clinical practice, allowing maximum efficacy with acceptable tolerability in individual patients. The objective of this analysis was to examine the dose-optimization process and the efficacy, safety, and tolerability of MPH XR-ODT in children 6–12 years of age with ADHD during the 5-week, open-label dose-optimization/stabilization period.
Methods
This is a secondary analysis of a phase 3, laboratory classroom study. Detailed methods for the classroom study have been published previously (Childress et al. 2017). The study was approved by a central Institutional Review Board (Copernicus Group, Research Triangle Park, NC) and conducted in accordance with Good Clinical Practice, IND regulations, and the most recent guidelines of the Declaration of Helsinki (ICH 1996; World Medical Association 2013). Written informed consent and assent were obtained from parents or legal guardians and participants, respectively, before performing any study-related procedures. Assent was obtained from all participants and documented by the child's signature, or as per local or site requirements.
Study procedures
All doses in the study are reported as the MPH hydrochloride (HCL) preparation dose equivalent (Engelking et al. 2018). After the washout period of 3–7 days, subjects underwent an open-label, stepwise dose-optimization period for 4 weeks (visits 2–5), starting at 20 mg per day and increasing each week (20 → 30 mg, 30 → 40 mg, and 40 → 60 mg per day) until an optimal dose was reached. An optimal dose of MPH XR-ODT produces a meaningful reduction in ADHD symptoms, (defined as a reduction of ≥30% according to the ADHD Rating Scale, Version IV [ADHD-RS-IV]), and a Clinical Global Impression-Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), while maintaining a tolerable AE profile. Participants remained on the optimized dose for 1 week (dose-stabilization period, visit 6 [beginning]–visit 7 [end]) before the randomized part of the study began. Daily doses of MPH XR-ODT were administered at home by a parent or legal guardian. MPH XR-ODT tablets were placed on the participant's tongue to allow the drug to disintegrate, and the disintegrated particles were swallowed. During the dose-stabilization period, participants continued to be evaluated for efficacy, safety, and tolerability.
Key inclusion criteria
Children (6–12 years of age) meeting the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM-IV, American Psychiatric Association 2000) criteria for ADHD (any subtype) were eligible. Participants had a positive response to previous treatment with a 20- to 60-mg daily dose of MPH for at least 1 month, CGI-Severity (CGI-S) score >3, and ADHD-RS-IV total or subscale score >90th percentile normative value for age and gender at baseline.
Key exclusion criteria
Children with a history of poor response, known allergy, or serious adverse reactions to any MPH formulation or components of MPH XR-ODT; with a history of another clinically significant psychiatric disorder; with a history of cardiovascular conditions; or requiring both a stimulant and nonstimulant to control ADHD were excluded. Other exclusion criteria were previously reported (Childress et al. 2017).
Efficacy and safety assessments
ADHD-RS-IV total and subscale scores were assessed at each visit. CGI-S was assessed at visit 2, followed by CGI-I assessment at each subsequent visit (visits 3–7). Safety was assessed with reported and observed treatment-emergent AEs (TEAEs), physical examinations, electrocardiograms, vital signs, clinical laboratory tests, and the Columbia-Suicide Severity Rating Scale (C-SSRS) Pediatric Baseline and Since-Last-Visit versions.
Statistical analysis
Final optimized MPH XR-ODT dose was defined as the assigned dose at the final dose-optimization period visit (visit 5). Final optimized dose (in mg and mg/kg) and number (%) of participants optimized at each dose (20, 30, 40, or 60 mg) were summarized for the total population and by sex, age range (6–7, 8–10, and 11–12 years), weight group (<30 kg, ≥30 kg), and baseline ADHD-RS-IV total score using descriptive statistics. A baseline ADHD-RS-IV total score of <42 for moderate symptom severity versus ≥42 for marked symptom severity was used based on an analysis linking ADHD-RS-IV total and CGI-S scores in children (6–12 years) with ADHD (Goodman et al. 2010).
The time to onset of TEAEs was measured as the number of days from first dose of MPH XR-ODT during the dose-optimization period. Duration of TEAEs was defined as number of days from AE start date to AE end date.
All analyses were performed in SAS®. Descriptive statistics were used to summarize ADHD-RS-IV total score; ADHD-RS-IV hyperactivity/impulsivity and inattentiveness subscale scores; CGI-I scores; the number (%) of participants at each dose level; and number (%) of participants whose MPH XR-ODT dose was increased, decreased, or maintained by time point. The least-squares (LS) mean ADHD-RS-IV total score by study visit was calculated post-hoc using a mixed-effects model for repeated measures with terms for final optimized dose, visit, the interaction of final optimized dose and visit, and baseline score.
Results
Participants
Eighty-seven participants (59 males and 28 females) were enrolled in the study; 85 completed dose optimization and 83 completed dose stabilization. Of the four participants who discontinued, two withdrew due to AEs (upper abdominal pain and influenza) and two withdrew consent. Of the 87 enrolled participants, most were 8–10 years old, 24.1% had inattentive type ADHD, 1.1% had hyperactive/impulsive type ADHD, and 74.7% had combined type ADHD. Participant demographics and clinical characteristics at baseline (visit 2) are summarized in Table 1.
Participant Demographics and Clinical Characteristics
ADHD, attention-deficit/hyperactivity disorder; ADHD-RS-IV, Attention-Deficit/Hyperactivity Disorder-Rating Scale-IV; BMI, body mass index; CGI-S, Clinical Global Impressions-Severity; max, maximum; min, minimum; MPH XR-ODT, methylphenidate extended-release orally disintegrating tablet; SD, standard deviation.
MPH XR-ODT dose optimization
All participants were given MPH XR-ODT 20 mg daily during the first study week, and the dose could be adjusted weekly. The distribution of participants at each dose by study week is shown in Figure 1. At visit 3, 24 participants remained on the 20-mg dose, while the daily dose was increased to 30 mg for 62 participants. At visit 4, the daily dose was 20 mg for 14 participants, 30 mg for 21 participants, and 40 mg for 51 participants. Also, at visit 5, the daily dose was 20 mg for 11 participants, 30 mg for 20 participants, 40 mg for 25 participants, and 60 mg for 30 participants. For the overall population, doses were maintained or increased during study weeks 3–5. When analyzed by age group, mean doses were increased for all groups at study visits 3–5. MPH XR-ODT dose was decreased at visit 6 (beginning of the dose-stabilization period) for two participants, one in age group 8–10 years and one in age group 11–12 years.
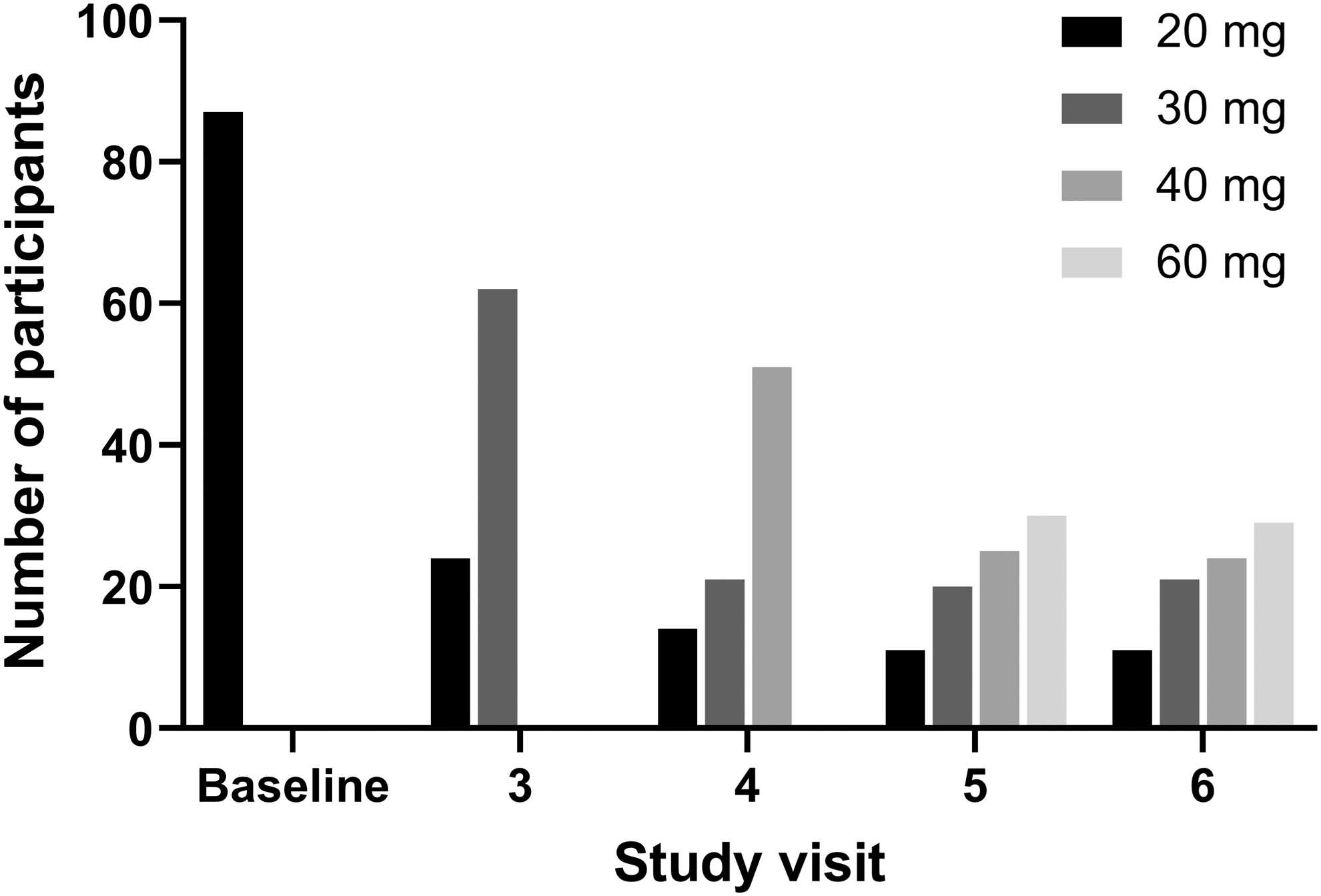
Distribution of individual MPH XR-ODT dose by study visit. MPH XR-ODT, methylphenidate extended-release orally disintegrating tablet.
The final optimized dose of once-daily MPH XR-ODT was 20 mg for 11 participants, 30 mg for 21 participants, 40 mg for 24 participants, and 60 mg for 29 participants (overall mean [standard deviation, SD] of 41.8 [14.6] mg; median 40.0 mg) (Table 2). Participants with a baseline ADHD-RS-IV score <42 received a lower optimal dose (mean [SD]: 37.1 [14.7] mg; median: 30.0 mg) than participants with a baseline ADHD-RS-IV score ≥42 (48.1 [11.9] mg; 40.0 mg) (Table 2). Participants with combined type ADHD received a higher optimized dose (mean [SD]: 43.8 [14.3] mg; median: 40.0 mg) than individuals with inattentive type (mean [SD]: 35.7 [14.3] mg; median: 30.0 mg). Mean optimal dose did not differ by age group or weight class, and median final dose did not differ by sex, age group, or weight class (all were 40.0 mg) (Table 2).
Final Optimized Dose of Methylphenidate Extended-Release Orally Disintegrating Tablet for the Overall Population and by Subgroup
Based on the third quartile of children who were moderately ill (CGI-S = 4) in Goodman et al. (2010).
ADHD, attention-deficit/hyperactivity disorder; ADHD-RS-IV, ADHD Rating Scale-IV; CGI-S, Clinical Global Impressions-Severity; max, maximum; min, minimum; MPH XR-ODT, methylphenidate extended-release orally disintegrating tablet; SD, standard deviation.
ADHD symptom assessment
Optimization of MPH XR-ODT was associated with a steady reduction in mean ADHD-RS-IV total score (Fig. 2A), ending with a mean change from baseline (SD) in ADHD-RS-IV score of −21.4 (8.9) at visit 7. ADHD-RS-IV inattention and hyperactivity/impulsivity subscale scores also improved from baseline to visit 7 (mean change from baseline [SD] of −11.5 [4.9] and −9.9 [5.7], respectively; Fig. 2B). Approximately 60% of participants achieved ≥50% improvement in ADHD-RS-IV total score from baseline.
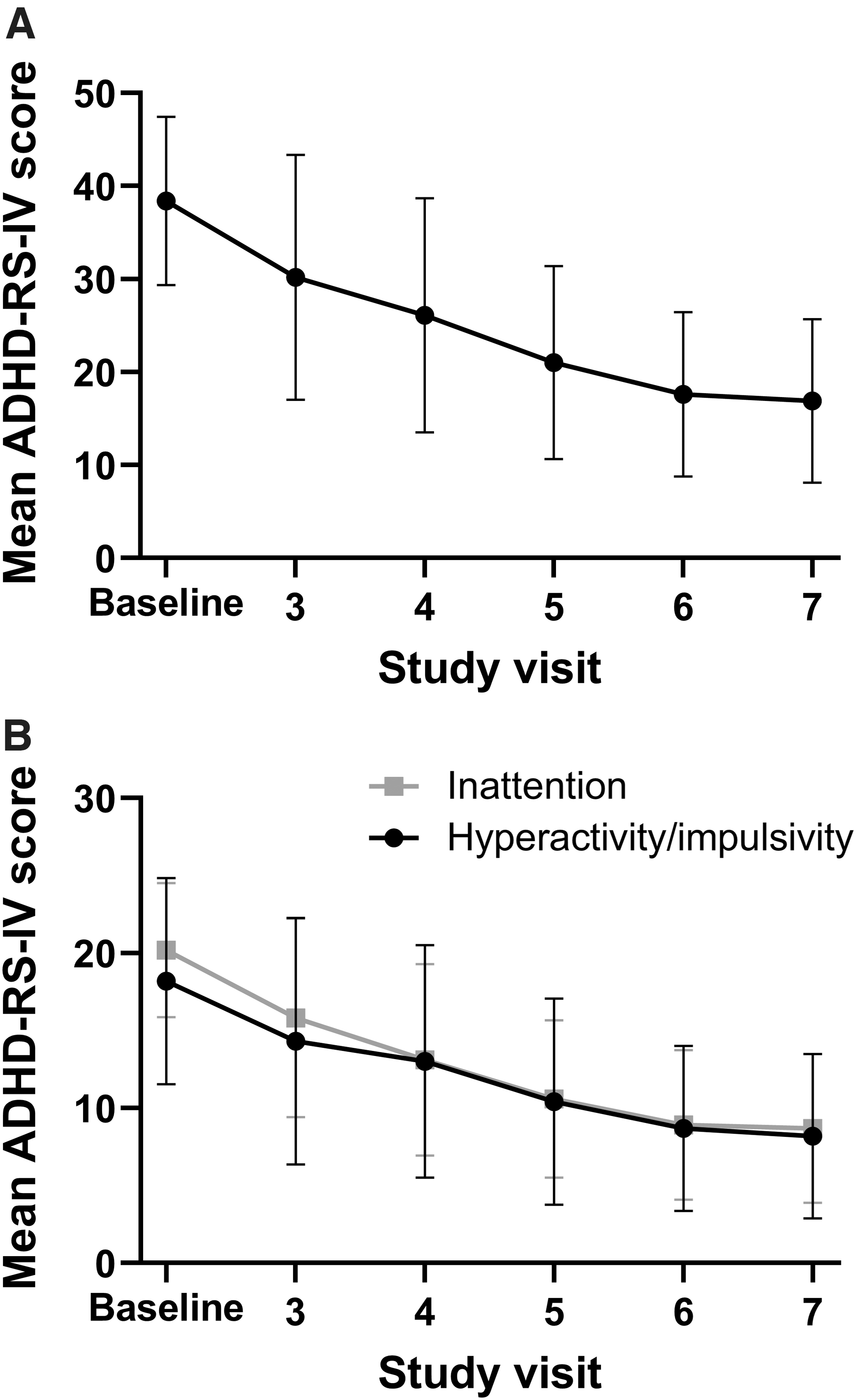
ADHD-RS-IV score during the dose-optimization and dose-stabilization period.
The pattern of improvement in ADHD-RS-IV total score over the dose-optimization period differed for participants based on the final optimized dose. ADHD-RS-IV total score at baseline was highest for participants optimized to the 40-mg dose of MPH XR-ODT (LS mean [standard error, SE]: 40.0 [1.4]) and lowest for those optimized to the 20-mg dose (34.8 [2.1]). Participants optimized to the 20-mg dose of MPH XR-ODT showed the most improvement within one study visit, with a mean (SD) change from baseline ADHD-RS-IV total score of −19.2 (7.7) at visit 3. However, participants optimized to the 60-mg dose experienced gradual improvement, beginning with a mean (SD) change from baseline in ADHD-RS-IV total score of −3.1 (6.3) at visit 3 and reaching −20.8 (8.6) at visit 6. Differences were observed in mean ADHD-RS-IV total score at visits 3 and 4 for all optimized dose groups (Fig. 3). The 20, 30, and 40 mg optimized dose groups reached a similar mean ADHD-RS-IV score by visit 5, and all optimized dose groups displayed a comparable mean ADHD-RS-IV score at visit 6 (Fig. 3). Age group, sex, and weight class comparisons did not affect ADHD-RS-IV score; however, when participants were compared by baseline ADHD-RS-IV score (<42 vs. ≥42), those with ADHD-RS-IV score ≥42 ended the dose-optimization period with a somewhat lower ADHD-RS-IV total score (18.8 vs. 16.0; Fig. 4).
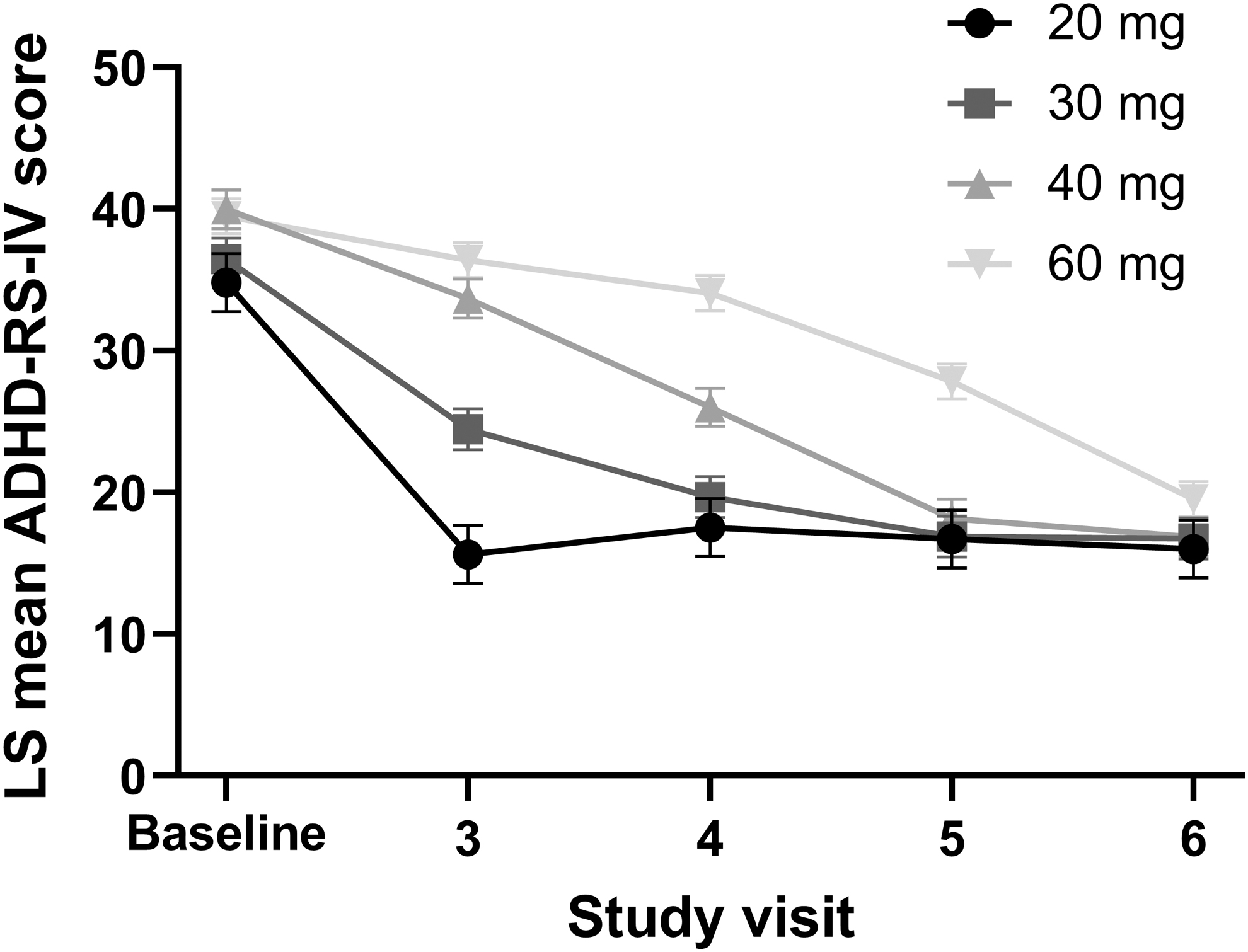
ADHD-RS-IV total score during the dose-optimization period by final optimized dose. Data displayed as LS mean and SE. ADHD-RS-IV total score was analyzed by visit using a mixed-effects model for repeated measures with terms for final optimized dose, visit, the interaction of final optimized dose and visit, and baseline score. ADHD-RS-IV, Attention-Deficit/Hyperactivity Disorder-Rating Scale-IV; LS, least squares; SE, standard error.
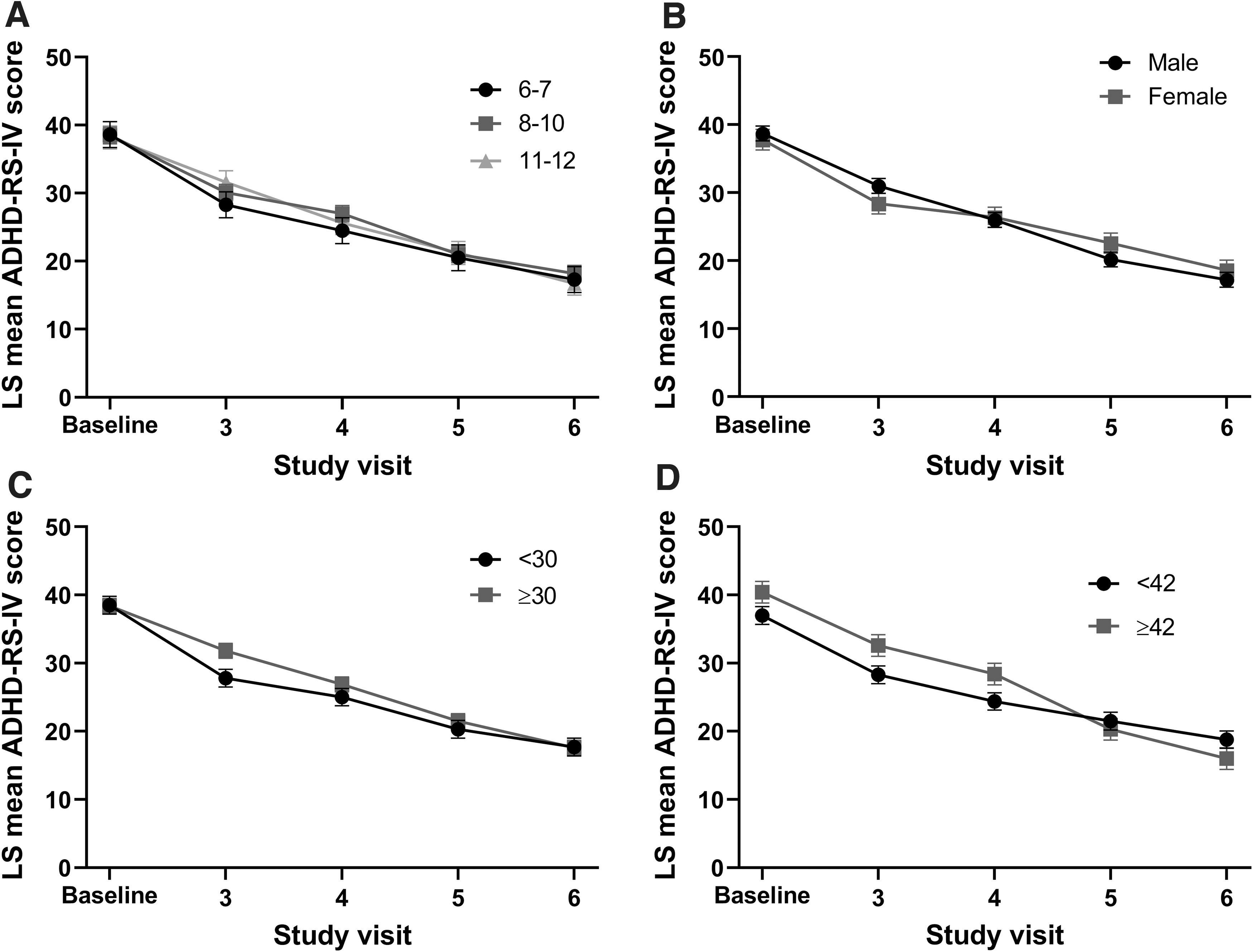
ADHD-RS-IV total score during the dose-optimization period by
At baseline (visit 2), CGI-S scores ranged from 4 (moderately ill) to 6 (severely ill). The mean (SD) CGI-S score was 4.5 (0.6) (Table 1). Over the course of the dose-optimization and dose-stabilization periods, mean (SD) CGI-I scores displayed steady improvement, shifting from minimally improved (3.1 [1.1]) at visit 3 to much improved (1.6 [0.6]) at visit 7.
Safety assessment
TEAEs were reported by 70/87 (80.5%) participants during the dose-optimization and dose-stabilization period. All TEAEs were mild or moderate; no serious AEs or deaths occurred. The most common TEAEs (>5% of participants) included decreased appetite, upper abdominal pain, headaches, and insomnia (Table 3). Time to onset was similar for decreased appetite (mean [SD]: 11.9 [11.5] days), upper abdominal pain (13.1 [10.0] days), headaches (14.9 [10.7] days), and insomnia (11.4 [10.1] days). Duration was longest for insomnia (25.4 [15.7] days) and decreased appetite (25.1 [12.8] days), while upper abdominal pain (14.1 [14.0] days), and headaches occurred for the shortest duration (3.2 [3.2] days). Suicidal ideation (Category 1: Wish to be dead) as per the C-SSRS was reported to have occurred one time for one participant at visit 5. The participant did not report suicidal ideation or behavior at any prior or subsequent visit and successfully completed the study. No suicidal behavior was reported during the dose-optimization period.
Treatment-Emergent Adverse Events in ≥5% of Participants During the Dose-Optimization and Dose-Stabilization Period
TEAE, treatment-emergent adverse event.
Discussion
ADHD treatment guidelines advise clinicians to perform dose optimization when starting a new pharmacotherapy (Pliszka and The AACAP Work Group on Quality Issues 2007; Wolraich et al. 2019). Treatment with the minimal prescribed dose may improve symptoms, but patients typically achieve better outcomes when treated with an individualized optimal dose. Dose optimization enables each patient to receive the most benefit from treatment while experiencing acceptable tolerability. However, a recent study of patients treated for ADHD at pediatric community practices of a large urban hospital found that the time to stable dose was >4 months for 25% of patients, with some patients taking several years to achieve a stable dose (Pasadyn et al. 2020).
Dose optimization is commonly performed during ADHD clinical trials (Childress et al. 2015, 2017; Huss et al. 2017; Wigal et al. 2018); however, there are a limited number of published studies reporting detailed analysis of efficacy endpoints after treatment with ADHD medication during the dose-optimization period for any age group (Huss et al. 2017; Wigal et al. 2018; Childress et al. 2020). In this study, we analyzed dose optimization of MPH XR-ODT in children with ADHD during the 5-week open-label portion of a classroom study. To easily compare this analysis with the original published classroom study and other studies of ADHD pharmacotherapy, we reported MPH XR-ODT doses as the HCL equivalents. Because of the technology used to create MPH XR-ODT, only the base MPH molecule is present, as opposed to the MPH HCL salt. Therefore, as per FDA guidelines, MPH XR-ODT is available as tablets of 8.6, 17.3, and 25.9 mg, which are equivalent to 10, 20, and 30 mg of MPH HCL (Engelking et al. 2018).
In this study, the optimal dose of MPH XR-ODT ranged from 20 to 60 mg, which is comparable to studies of other MPH formulations in children 6–12 years of age with ADHD, which show participants were optimized to all available doses (Wigal et al. 2013, 2018). The mean optimized MPH XR-ODT dose (41.8 mg) was also similar to the mean final optimized dose reported for a dose-optimization analysis of MPH extended-release chewable tablets (42.6 mg; ERCT) (Wigal et al. 2018).
The pattern of symptom improvement varied for each optimized dose group. Participants optimized to the 20 mg/day dose achieved greater improvement in ADHD symptoms within 1 week of MPH XR-ODT treatment, whereas those with a higher final optimized dose showed gradual ADHD symptom improvement as their doses were increased to the final optimized dose. However, by the end of the 4-week dose-optimization period, the mean ADHD-RS-IV total score was similar for all optimized dose groups. This finding was comparable to previously published dose-optimization results for MPH ERCT (Wigal et al. 2018), further confirming that dose optimization is needed for some patients to achieve optimal response to treatment.
In this study, the MPH XR-ODT mean optimal dose was similar regardless of age group, which contrasts with the study for MPH ERCT where mean optimized dose increased with age (Wigal et al. 2018). However, the median optimized dose of 40 mg for all age groups was consistent between the two studies (Wigal et al. 2018). Weight class and sex did not have an effect on outcomes of dose optimization in either study (Wigal et al. 2018), although both studies found a relationship between baseline ADHD-RS-IV total score and final optimized dose (Wigal et al. 2018). Participants with higher ADHD-RS-IV baseline scores were optimized to a higher final dose of MPH XR-ODT compared to participants with lower baseline scores. A higher final dose was also needed for participants with combined type ADHD. ADHD subtype may influence ADHD severity; children and adolescents with combined type ADHD had greater symptom severity at baseline (Zhang et al. 2005). The data suggest that higher dosing within labeling guidelines in the clinic could provide greater symptom improvement for children 6–12 years of age.
MPH XR-ODT was well tolerated during the study. The safety profile of MPH XR-ODT was consistent with that of other extended-release MPH formulations, which is a mixture of mild, moderate, and severe TEAEs commonly, including decreased appetite, upper abdominal pain, affect lability, insomnia, irritability, and headache (Murray et al. 2011; Wigal et al. 2013). Notably, only two patients discontinued due to AEs and only two required dose decreases in this study of MPH XR-ODT.
Limitations
This was a secondary analysis of the open-label phase of a laboratory classroom study. The original study aimed to optimize dosage before a double-blind treatment phase for the classroom study; therefore, this analysis lacks a placebo control against which to compare efficacy and safety results. This analysis could also benefit from a control group in which the 20-mg dose was continued for the duration of the study, allowing an efficacy comparison between dose optimization and the minimum prescribed dose. Inclusion criteria required participants to have been on a stable dose of MPH medication for 1 month before washout; therefore, the children were already tolerant to MPH.
Conclusion
Although MPH dose is titrated weekly in clinical trials, this rarely occurs in clinical practice, where achieving a stable dose of medication or maximum symptom improvement can take 3–4 months or longer (Epstein et al. 2010; Pasadyn et al. 2020). These data suggest that patients with more severe ADHD symptoms can benefit from weekly titration to an optimal dose while monitoring tolerability.
Clinical Significance
MPH XR-ODT treatment during the open-label period was associated with improvement in ADHD symptoms, as indicated by a clinically significant decrease in both ADHD-RS-IV total and CGI-I and subscale scores. This study highlights the individual variability in dose–response as participants were optimized to every available dose. Higher baseline ADHD-RS-IV total score corresponded with a higher final optimized dose of MPH XR-ODT. This dose-optimization analysis revealed a relationship between optimal dose and change in ADHD symptoms, which can help guide clinicians in optimizing stimulant dose based on baseline severity.
Footnotes
Acknowledgments
This research was supported by Neos Therapeutics, Inc., Grand Prairie, TX. Writing and editorial support were provided by Shavonn Harper, PhD, and Nicole Seneca, PhD, of AlphaBioCom, LLC, King of Prussia, PA.
Disclosures
A.C.C. has received research support from, consulted with, acted as an invited speaker for, and/or served on advisory boards for Adlon Therapeutics; Akili Interactive; Alcobra Pharma; Allergan; Arbor Pharmaceuticals; Cingulate Therapeutics; Emalex; Forest Research Institute; Ironshore Pharmaceuticals; Jazz; KemPharm, Inc.; Lundbeck; Neos Therapeutics; NLS Pharma; Otsuka Pharmaceutical; Pfizer; Purdue Pharma; Rhodes Pharmaceuticals; Shire Pharmaceuticals; Sunovion Pharmaceuticals; Supernus Pharmaceuticals; Takeda; Tris Pharma; and the United States Food and Drug Administration.
S.H.K. has received research support from and/or consulted with Akili Interactive; Alcobra Pharma; Arbor Pharmaceuticals; Atentiv; National Institutes of Health (NIDA, NIEHS, NICHD); Neos Therapeutics; Neurovance; Purdue Pharma; Rhodes Pharmaceuticals; Shire Pharmaceuticals; Sunovion Pharmaceuticals; Supernus Pharmaceuticals; Tris Pharma; and the US Environmental Protection Agency.
A.J.C. is an employee and board member of the Neuroscience Education Institute and has received research support from, consulted with, and/or acted as invited speaker for Adlon Therapeutics, Aevi Genomics, Akili Interactive, Arbor Pharmaceuticals, Atentive, KemPharm, Ironshore Pharmaceuticals, Neos Therapeutics, Noven Pharmaceuticals, Otsuka, Purdue Pharma, Shire Pharmaceuticals, Sunovion Pharmaceuticals, Supernus Pharmaceuticals, Takeda Pharmaceuticals, and Tris Pharma.
A.M. has received research support from, consulted with, and/or acted as an invited speaker for Acadia Pharmaceuticals; Akili Interactive Labs; Allergan; Arbor Pharmaceuticals, LLC; Avanir; Boehringer Ingelheim Pharmaceuticals, Inc.; Eisai, Inc.; Ironshore Pharmaceuticals & Development, Inc.; KemPharm, Inc.; Neos Therapeutics; Novartis Pharmaceuticals Corporation; Otsuka America Pharmaceutical, Inc.; Purdue Pharma; Roche; Sage Therapeutics; Shire; Sunovion Pharmaceuticals, Inc.; Supernus Pharmaceuticals, Inc.; Takeda Pharmaceutical Company Ltd.; Tonix Pharmaceuticals; and Tris Pharma.
C.R.S. was an employee of Neos Therapeutics, Inc., at the time this study was conducted, and holds stock in Neos Therapeutics and in Pfizer, Inc.