
Abstract
Background:
Placebo response has been identified as an important factor influencing the success of adult antidepressant trials, yet little research of placebo response has been conducted in pediatric populations. Understanding disorder-specific and transdiagnostic predictors of pediatric placebo response is important in designing successful child psychopharmacological trials.
Methods:
A PubMed search was conducted for all pediatric antidepressant randomized controlled trials treating depression, anxiety, or obsessive-compulsive disorder (OCD). A random-effects model was utilized to examine the magnitude of placebo symptom improvement using standardized mean difference (SMD) and placebo response rates. Stratified subgroup analysis was performed by diagnostic indication. Meta-regression was utilized to search possible correlates of placebo symptom improvement and placebo response rate.
Results:
Thirty antidepressant trials involving 2911 participants receiving placebo were included in this meta-analysis. Magnitude of placebo improvement and placebo response rates varied significantly across disorders; being greater in depression (SMD = 1.44, 95% confidence interval [CI]: 1.18 to 1.71) than anxiety disorders (SMD = 1.09, 95% CI: 0.77 to 1.41) and the lowest in OCD (SMD = 0.71, 95% CI: 0.32 to 1.12). Different predictors were associated with placebo response in different indications.
Conclusions:
Both the magnitude and predictors of placebo response in pediatric depression trials do not replicate across anxiety and OCD. Based on our results, across disorders, minimizing the number of sites might significantly reduce placebo improvement. In addition to these, we could potentially decrease the placebo response in depression trials by increasing the number of subjects enrolled per study site, minimizing the number of study visits and conducting the studies in the United States. Further research is needed into the predictors of placebo response in pediatric anxiety and OCD.
Introduction
Depressive disorder, anxiety disorders, and obsessive-compulsive disorders (OCDs) are among the most prevalent psychopathologies in children and adolescents. They are disabling and are associated with adverse long-term outcomes; therefore, early diagnosis and treatment are critical. Selective serotonin receptor inhibitors (SSRIs) and serotonin–norepinephrine receptor inhibitors (SNRIs) are the first- and second-line pharmaceutical treatments for these disorders (Locher et al. 2017) despite the fact that their efficacy has been much debated (Thase 2002; Kirsch 2014b). The controversy stems from the relatively small difference in effect size between placebo and active drugs in randomized control trials (RCTs) (Mancini et al. 2014).
Over the past 30 years, antidepressant RCTs have shown a trend of decreasing efficacy (Khin et al. 2011; Rutherford and Roose 2013) with roughly half of the RCTs in both adults and children failing to show any significant difference from placebo (Khan et al. 2003b; Gelenberg et al. 2008; Turner and Rosenthal 2008; Khin et al. 2011). These failures have been demonstrated to be owing to the large and steadily rising placebo response observed in antidepressant trials (Rutherford and Roose 2013; Mancini et al. 2014). This high placebo response reduces signal detection resulting in delay or failure of psychiatric drug discovery and has raised doubts regarding the effectiveness of SSRIs and SNRIs (Kirsch 2014a).
In adult antidepressant trials, the size of the placebo response has been found to be the single most influential factor that determines the outcome of a trial (Khan et al. 2003a) and yet only 2.5% of the literature on the placebo effect pertains to the pediatric population (Weimer et al. 2013). This is owing to reduced number of psychopharmacological trials conducted in children, as well as ethical and logistical concerns conducting research examining placebo response (a.k.a. having “no treatment” or a “no treatment” arm) in children (a protected population in research (Koechlin et al. 2018). The extent that the findings in adult antidepressant trials can be extrapolated into the pediatric population is unknown. Children differ from adults with regard to their physiology, pathophysiology, pharmacokinetics, pharmacodynamics, neural development and possibly placebo response (Weimer et al. 2013). In addition, children interact within a different environment than adults, therefore, effects such as “placebo by proxy” (the influence of parental expectations or increased attention from doctors and caregivers) could have a greater influence on their response (Weimer et al. 2013). Thus, identifying potential predictors specific to the pediatric placebo response in internalizing disorders is crucial in aiding the development of pediatric psychopharmacology. In addition, this information can allow clinicians to maximize the placebo effect in the therapeutic setting (Mintz and Flynn 2012).
Both Mayes et al. (2007) and Bridge et al. (2009) found that the placebo response in depression trials was higher in children younger than the age of 12 compared with adolescents. Bridge et al. (2009) reviewed 12 pediatric depression trials and reported a greater number of study sites to be the greatest predictor of a higher placebo response. In addition, greater baseline severity of depressive symptoms was associated with lower rates of placebo response. Similarly, Strawn et al. (2017) examined the placebo response in pediatric anxiety and found separation anxiety to be the strongest predictor of placebo response. Treatment expectation was also correlated to a higher placebo response, with placebo response being greater early during treatment and plateauing by week 8. As mentioned, there is more literature on the placebo response in the adult populations, which reports being female and non-Caucasian, having increased trial duration, shorter illness duration, less recurrent episodes, non-U.S. study sites, and earlier date of publication decreases the placebo response (Khan et al. 1991, 2007; Wilcox et al. 1992; Charney et al. 2002; Walsh et al. 2002; Stein et al. 2006; Posternak and Zimmerman 2007; Kirsch et al. 2008).
Although different antidepressant agents appear to have similar efficacy in pediatric populations across all three diagnostic groups, meta-analyses have shown efficacy to be the greatest for anxiety, intermediate for OCD, and least for depression, with numbers needed to treat being 3, 6 and 10, respectively (Bridge et al. 2009). Cohen et al. (2010) demonstrated that differences in efficacy in pediatric antidepressant trials are owing to a greater placebo response in studies on depression, compared with those on anxiety and OCD (49.6% [range: 17%–90%] vs. 39.6% [range: 9%–53%] vs. 31% [range: 4%–41%], respectively, ANOVA F = 7.1, p = 0.002). To date, several meta-analyses have examined other predictors of placebo response within pediatric depression and anxiety disorders separately; however, none have compared it across diagnoses (Bridge et al. 2009; Cohen et al. 2010; Strawn et al. 2017). It may be that the differences across diagnosis result from certain symptoms being more susceptible to the placebo response. Taking both trans-diagnostic and categorical approaches to study trial and subject factors associated with placebo response within and across diagnoses will possibly increase our understanding in treating these conditions and lead to better pediatric clinical trial design.
This meta-analysis studies the placebo effect in all the available double-blinded, randomized, placebo-controlled SSRI and SNRI trials conducted on the pediatric population with the primary diagnosis of depression, anxiety disorders, or OCD. It examines symptom improvement and response rates across these trials and investigates potential trial and subject-level predictors of the placebo response, both within and across the disorders. Understanding which predictors are disorder specific and which predictors are transdiagnostic will help in the design of future pediatric trials examining internalizing disorders.
Methods
Search strategy for identification of relevant studies
This study was conducted according to the Preferred Items for Systematic Reviews and Meta-Analyses (PRISMA) statement (Liberati et al. 2009; Moher et al. 2009). PubMed was searched by two reviewers (M.N. and J.L.) to identify eligible studies (last search conducted on May 14, 2020). The search terms used were (“Anxiety Disorders”[Mesh] OR “Depressive Disorder”[Mesh] OR “Obsessive-Compulsive Disorder”[Mesh]) AND “Randomized Controlled Trial” [Publication Type] AND “Serotonin Uptake Inhibitors”[Mesh] AND (“Child”[Mesh] OR “Adolescent”[Mesh]). Results of pharmacological trials were restricted to RCTs, meta-analyses, and reviews. The references of the included studies, meta-analyses, and reviews were examined for additional trials (Bridge et al. 2007, 2009; Strawn et al. 2017). The protocol was not preregistered before conducting the search or data analysis.
Criteria for inclusion and exclusion of studies in the meta-analysis
The search titles and abstract obtained by this search strategy were uploaded to the online Rayyan QCRI application (“Rayyan QCRI, the Systematic Reviews web app,” 2016) for systemic reviews and examined independently by two reviewers (M.N. and S.T.) to determine inclusion in this meta-analysis. Any discrepancies were resolved by a final reviewer (M.H.B.). The following criteria were used to determine inclusion or exclusion.
Types of studies
Studies were included in the meta-analysis if they were randomized, double-blinded, placebo-controlled SSRI and SNRI clinical trials. Case reports, letters, and reviews were excluded. RCTs were excluded if they were discontinuation trials or secondary analyses of already included trials.
Types of participants
Children younger than the age of 18 with a current primary psychiatric diagnosis of any indication approved for antidepressant pharmacotherapy. These included depression, anxiety disorders, and OCD. Comorbidity was allowed and information about comorbidity was extracted.
Types of settings
The study included trials conducted at both academic and nonacademic sites, nationally and internationally. Owing to limited resources, only studies published in English were included.
Types of intervention
Trials needed to have two interventions to be included, the placebo pill and an SSRI/SNRI antidepressant. We a priori chose to exclude trials of tricyclic antidepressants, mirtazapine and bupropion, as they have increased prevalence of side-effects compared with SSRI/SNRIs and thus are much more likely to functionally unblind trials altering the placebo response. In addition, there are extremely few studies of these medications. We also chose not to include placebo-controlled trials of antipsychotic medications (of which there are many randomized placebo-controlled trials in pediatric populations) because of the same issues with functional unblinding of placebo arms and that these medications are in general used for very different indications (bipolar disorder, psychosis, and aggression) and over shorter durations of treatment. Studies were excluded if the placebo or antidepressant was not given in pill form. Studies were excluded if their placebo group received an additional alternative treatment such as cognitive behavioural therapy (CBT) (Melvin et al. 2017).
Types of outcome measures
The study needed to compare SSRI/SNRI pharmacotherapy to placebo using well-validated measures that applied the Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria to measure symptoms. These measures include the Children's Depression Rating Scale–Revised (CDRS-R) (Isa et al. 2014), Montgomery–Asberg Depression Rating Scale (MADRS) (Davidson et al. 1986), Hamilton Depression Rating Scale (HAM-D) (Miller et al. 1985) for depression; Hamilton Anxiety Rating Scale (HAM-A) (Zimmerman et al. 2019), Pediatric Anxiety Rating Scale (PARS), Kiddie Schedule for Affective Disorders and Schizophrenia (K-SADS) (Axelson et al. 2003) for anxiety disorders and Children's Yale-Brown Obsessive-Compulsive Scale (CY-BOCS) (Storch et al. 2019) for OCD.
Studies with missing data were excluded, for example, if they only reported general severity and improvement scales (such as the Clinical Global Impression–Severity [CGI-S] or Clinical Global Impression–Improvement [CGI-I]) (Busner and Targum 2007) or if they did not provide pre- and postintervention data or mean change. The two outcome measures utilized were mean difference between baseline and endpoint ratings as well as the categorical response rates in the placebo arms of the trials.
Data extraction
Data from eligible studies was extracted independently by reviewers (M.N. and S.C.) and entered using Microsoft Excel 2010. Discrepancies were resolved by consensus. In addition to the outcome measures, variables were extracted to be examined as potential predictors from each study based on previous evidence (Rutherford and Roose 2013). These included the following: number of recruitment sites, proportion of academic sites, publication year, study duration, placebo lead in, number of study visits, samples size, randomization to active arm, subjects per site, United States versus international sites, funding source, mean age, percentage female, percentage Caucasian, and baseline severity. Unique information about each study was also noted.
If the study included multiple outcome measures for the same indication, then only the primary outcome measure was extracted. Outcome measures based on intent-to-treat (ITT) analysis were prioritized. If the study reported a second active SSRI/SNRI comparator arm, this additional arm was entered as a separate study compared with the placebo arm. If the study had a double-blinded extension phase, for long-term effects of the drug, this phase was entered as a separate study with separate time points. If the study pooled data from more than one trial, data from each included trial was entered separately to be treated independently for analysis (Emslie et al. 2007; Rynn et al. 2007). If the study was a crossover trial, outcome measures were only taken from the endpoint of the first phase of the study, before the crossover (Riddle et al. 1992). When standard deviations were not reported, these were calculated from standard errors, t values or p values that related to the differences between mean values in the two groups. Two studies (Wagner et al. 2004b, 2006) did not include any of these values and standard deviations were imputed by the “leaving one out method” (Furukawa et al. 2006). If trial outcomes were reported in a figure (Wagner et al. 2003), a graph digitizer program called GetData (“Digitize graphs and plots–GetData Graph Digitizer–graph digitizing software,” 2013) was used to extract the mean change from baseline.
Statistical procedures and conventions
Comprehensive Meta-Analysis V3 (Biostat, Englewood, NJ) was used for calculations and analyses.
Effect size computation
Primary outcome analysis was based upon “intention-to-treat” data from the completion of RCTs, before any crossover or open-label extension. Three effect sizes were calculated for each included study. First, the placebo effect was assessed as the mean change between baseline and endpoint scores and analyzed using standardized mean differences (SMDs). SMD is used to control for comparisons of studies utilizing different measures to assess the same outcomes of interest. The SMD is obtained by dividing the mean change by the standard deviation of outcome among participants. A secondary outcome examined was proportion of responders to placebo in trials. The criteria for treatment response were based on the definition set forth in individual trials.
Assessing heterogeneity
Heterogeneity was assessed by calculating the Q statistic (Cochran 1950), and the I 2, a transformation of Q that indicates the proportion of observed variance that can be attributed to heterogeneity rather than sampling error (Higgins et al. 2003). The Q statistic and has a chi-squared distribution with degrees of freedom equal to the number of studies minus 1.
Publication bias
Publication bias was assessed by creating funnel plots (Duval and Tweedie 2000) for the outcome measures by plotting the effect size against standard error for each included trial. In addition, publication bias was statistically tested by the Egger test (Egger et al. 1997) and by determining the association between sample size and effect size in meta-regression.
Selection of type of effects model
We chose to use a random-effects model as the primary model for meta-analysis because we expected substantial heterogeneity between trials and that differences between trials (not simply sampling error) would be associated with placebo response.
Subgroup and predictor analysis
The test for subgroup differences was used to examine whether diagnostic indication (depression, anxiety, or OCD) had a significant effect on symptom improvement.
Predictor analyses were conducted for the aforementioned continuous and categorical variables. These were initially run by performing meta-regression including just the variable of interest and then adjusted for diagnostic indication. We adjusted primary models for diagnostic indication as we believed the primary indication was an important predictor of placebo response and also likely correlated with other moderating variables of interest (Higgins et al. 2003).
Results
Included studies
A total of 4899 references were identified in PubMed. A total of 30 trials were eligible for potential inclusion once the titles, abstracts, and full texts were screened. One study (Simeon et al. 1990) did not provide placebo data for contribution to the meta-analysis outcomes. Our meta-analysis involved data from 30 trials involving 2911 subjects contributing to the placebo symptom improvement outcome and 26 trials with 2562 subjects contributing to the placebo response rate outcome. Supplementary Figure S1 provides a PRISMA diagram explaining our selection of trials. The characteristics of the included trials are given in Table 1.
The Characteristics of Included Trials
N included in our analysis. Some studies included additional arms (tricyclic antidepressant, CBT alone, antidepressant+CBT, etc.) that were not extracted.
Based on adequacy of random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, usage of observer-rated outcomes, completeness of outcome data, specification of main outcomes a priori, reporting on all outcomes, and conduction of ITT analyses. Individual items were summed and divided by the total number of applicable items to produce a total score ranging from 0 to 1, where higher scores denote greater methodological quality.
Only SSRI or SNRI treatment arms from acute treatment phase/precrossover phase extracted as per protocol.
Range. Mean (SD) not available.
Study medication and matching placebo provided by the drug manufacturer.
Two trials. Analyses broken down by study.
CBT, cognitive behavioural therapy; CDRS-R, Children's Depression Rating Scale–Revised; CY-BOCS, Children's Yale-Brown Obsessive-Compulsive Scale; DDNOS, depressive disorder not otherwise specified; Dx, diagnosis; GAD, generalized anxiety disorder; HAM-A, Hamilton Anxiety Rating Scale; HAM-D, Hamilton Depression Rating Scale; ITT, intent-to-treat; K-SADS, Kiddie Schedule for Affective Disorders and Schizophrenia; MADRS, Montgomery–Asberg Depression Rating Scale; MDD, major depressive disorder; NR, not reported, OCD, obsessive-compulsive disorder; PARS, Pediatric Anxiety Rating Scale; PD, panic disorder; PH, specific phobia; SAD, separation anxiety disorder; SD, standard deviation; SM, selective mutism; SMD, standardized mean difference; SNRI, serotonin–norepinephrine receptor inhibitor; SP, social phobia; SSRI, selective serotonin receptor inhibitor; SUD, substance use disorder; Tx Dur, treatment duration.
Quality analysis was conducted on the adequacy of randomization, blinding, allocation concealment, blinding of outcome assessment, using observer-rated outcomes, specifying main outcomes a priori, reporting on all outcomes, and conducting ITT analysis. Most of the trials were of high quality; all of them being double blinded and utilized random allocation. According to the Cochrane Risk of Bias Assessment Tool, there was a large amount of high risk in the “other risk of bias” category, which was mainly because of per protocol analysis rather than ITT analysis. In addition, the fact that the outcome measures being investigated do not include the primary outcomes of the studies (i.e., the effect of the active drug on disease) decreased the potential for bias.
Placebo symptom improvement
Thirty trials involving 2911 participants receiving placebo contributed to this outcome (Riddle et al. 1992, 2001; Emslie et al. 1997, 2002, 2006, 2007, 2009, 2014; March et al. 1998, 2004, 2007; Geller et al. 2001, 2004; Keller et al. 2001; Rynn et al. 2001, 2007; Walkup et al. 2001, 2008; Liebowitz et al. 2002; Birmaher et al. 2003; Wagner et al. 2003, 2004a, 2006; POTS 2004; Berard et al. 2006; Findling et al. 2009; da Costa et al. 2013; Atkinson et al. 2014; Strawn et al. 2015). The overall placebo symptom improvement from the baseline in the included trials was SMD = 1.21 (95% confidence interval [CI]: 1.15 to 1.27, z = 39.27, p < 0.01). There was significant heterogeneity in placebo symptom improvements between studies (I 2 = 90%, Q = 222, df = 27, p < 0.001) but there was no evidence of publication bias (Egger's regression: intercept = −1.05, 95% CI: −4.79 to 2.68, p = 0.28). The test for subgroup differences demonstrated that there was a significantly different symptom improvement based on diagnostic indication (test for subgroup differences χ2 = 67.6, df = 2, p < 0.001). The greatest symptom improvement on placebo was observed in trials of depression (SMD = 1.44, 95% CI: 1.18 to 1.71, z = 10.55, p < 0.01 and heterogeneity I 2 = 87%, Q = 69.3, df = 9, p < 0.001) compared with anxiety disorders (SMD = 1.09, 95% CI: 0.77 to 1.41, z = 6.74, p < 0.01 and heterogeneity I 2 = 91.9%, Q = 147.7, df = 12, p < 0.001) and OCD (SMD = 0.71, 95% CI: 0.32 to 1.12, z = 3.54, p < 0.01 and heterogeneity I 2 = 0%, Q = 5.46, df = 6, p < 0.486). Figure 1A provides a forest plot that demonstrates the symptom improvement with placebo in individual trials stratified by diagnostic indication.
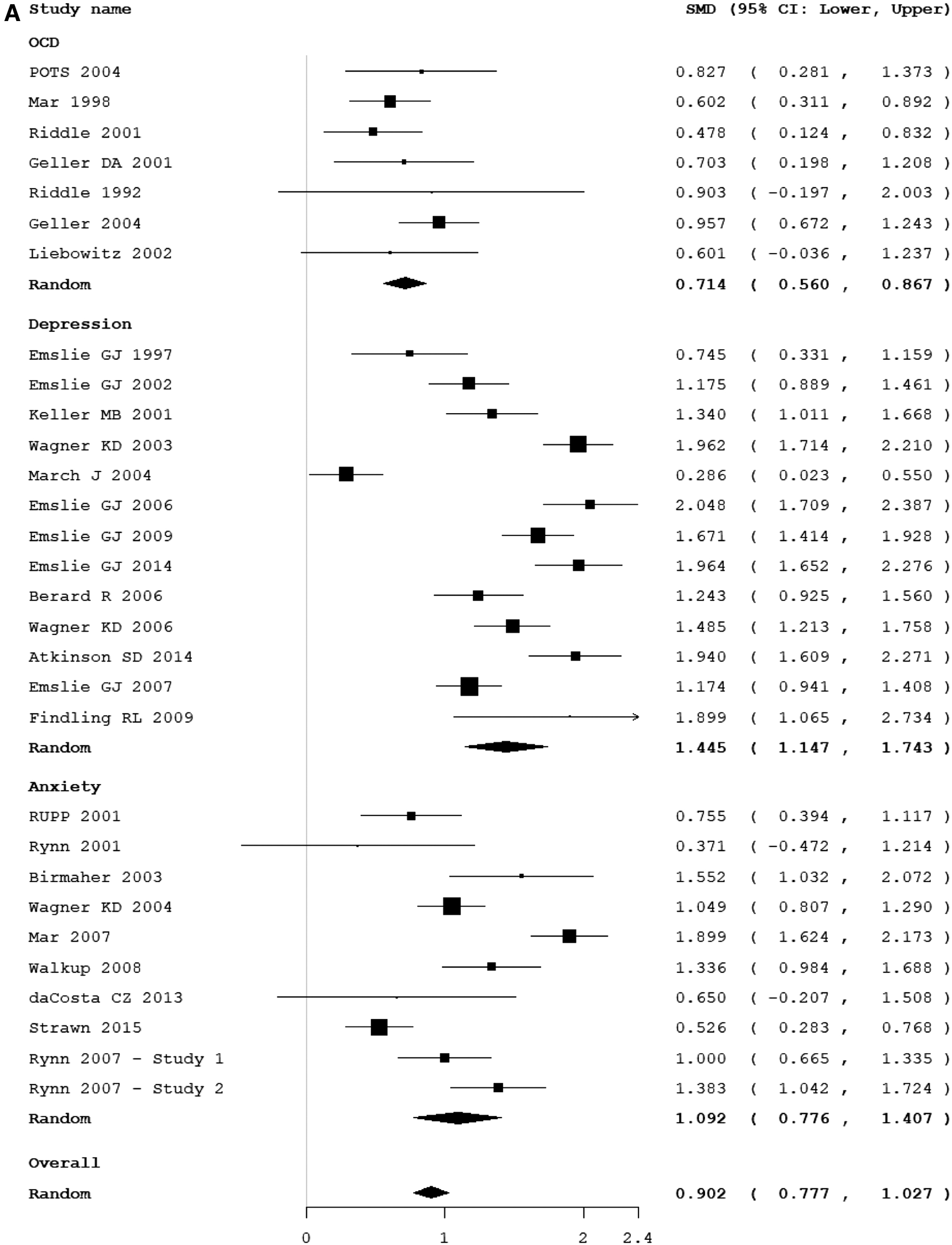
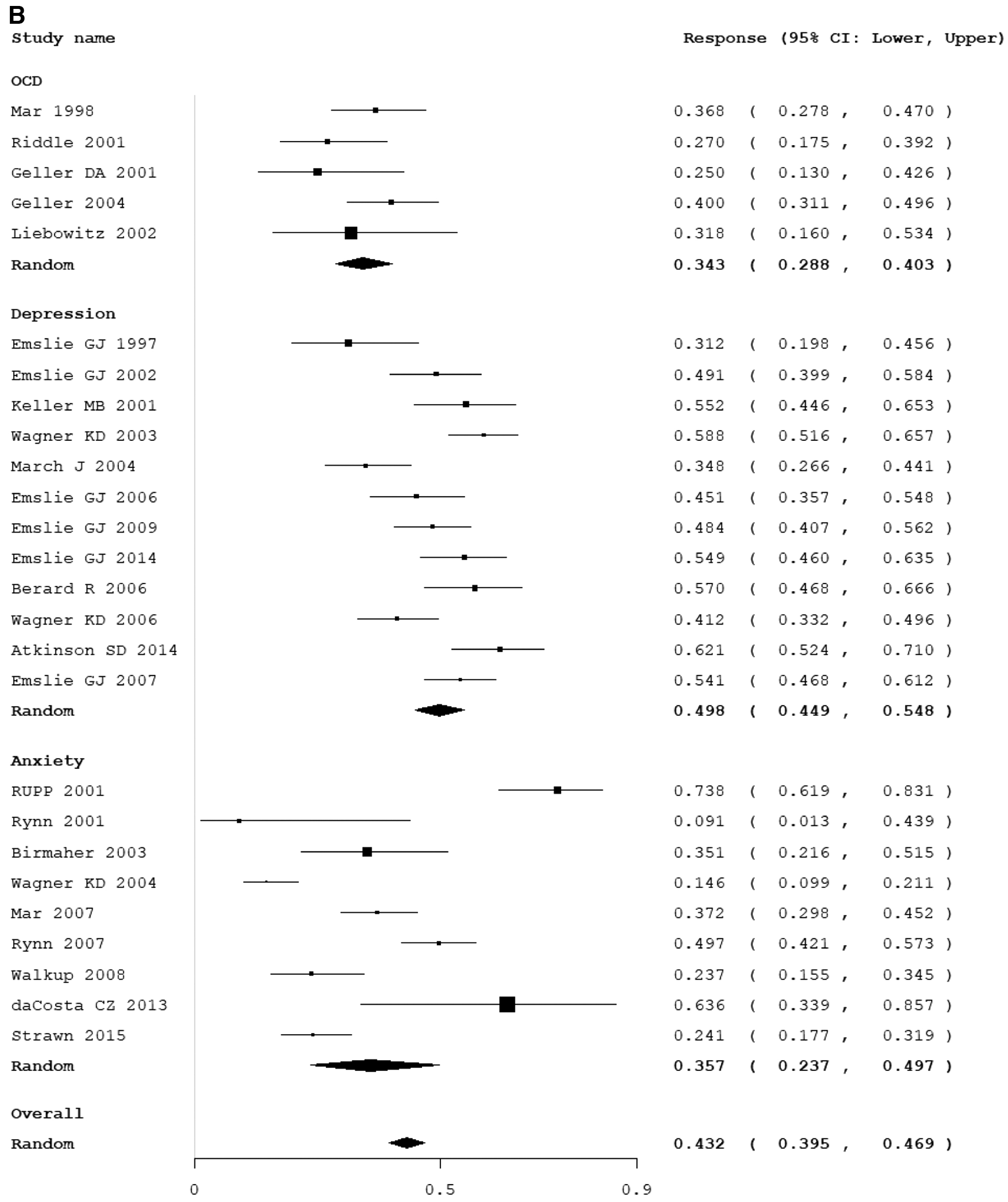
Table 2 provides the results of meta-regression analysis both within and across diagnostic indications. Across all disorders more study sites, international sites remained significantly associated with higher placebo after controlling for indication. Examining subject characteristics, a lower percentage of Caucasians was associated with higher symptom improvement in the placebo group. Among OCD trials specifically, the presence of a placebo lead-in phase and increasing duration of the placebo lead-in phase were associated with decreasing placebo symptom improvement. OCD trials conducted in the United States had a significantly lower placebo symptom improvement than trials conducted in other countries. Among pediatric depression trials, later year of publication, more study sites and a lower number of subjects per study site were associated with greater placebo symptom improvement. No predictors were statistically significant when exploring anxiety trials.
Meta-Regression Analysis of Predictors of Symptom Improvement
Table displays results of meta-regression of trial level and subject level predictors of symptom improvement with values of PE, upper and lower CI and p-value. Statistically significant results (p = <0.05) are in bold.
Randomization to active is the ratio of participants randomized to the active group to the placebo group.
Primarily industry funding indicates the source of the bulk of funding for the study.
Mean active dose is the average dose across all the participants in the active group reported by the study.
CI, confidence intervals; OCD, obsessive-compulsive disorder; PE, point estimates; SSRI, selective serotonin receptor inhibitor.
Placebo response rate
Twenty-six trials involving 2562 participants in the placebo group defined the criteria for response and reported the response rate (Emslie et al. 1997, 2002, 2006, 2007, 2009, 2014; March et al. 1998, 2004, 2007; Geller et al. 2001, 2004; Keller et al. 2001; Riddle et al. 2001; Rynn et al. 2001, 2007; Walkup et al. 2001, 2008; Liebowitz et al. 2002; Birmaher et al. 2003; Wagner et al. 2003, 2004a, 2006; Berard et al. 2006; Findling et al. 2009; da Costa et al. 2013; Atkinson et al. 2014; Strawn et al. 2015). The overall placebo response rate was 0.44 (95% CI: 0.43 to 0.47, z = −5.3, p < 0.01). There was significant heterogeneity in placebo symptom improvements between studies (I 2 = 87%, Q = 100.08, df = 25, p < 0.01) but there was no evidence of publication bias (Egger's regression: intercept = −2.70, 95% CI: −6.02 to 0.621, p = 0.053). The test for subgroup differences demonstrated that there was a significantly different placebo response rate based on diagnostic indication (test for subgroup differences χ2 = 16.41, df = 2, p < 0.001). The highest response rate was observed in trials of depression (event rate = 0.498, 95% CI: 0.449 to 0.548, z = −0.072, p = 0.943 and heterogeneity I 2 = 91.3%, Q = 92.4, df = 8, p < 0.001) compared with anxiety disorders (event rate = 0.357, 95% CI: 0.237 to 0.497, z = −2.003, p = 0.045 and heterogeneity I 2 = 91.9%, Q = 147.7, df = 12, p < 0.001) and OCD (event rate = 0.343, 95% CI: 0.288 to 0.403, z = −5.007, p < 0.001 and heterogeneity I 2 = 11.3%, Q = 4.51, df = 4, p < 0.341). Figure 1B provides a forest plot that demonstrates the response rate with placebo in individual trials stratified by diagnostic indication.
Table 3 provides the results of meta-regression analysis both within and across diagnostic indications for placebo response. There were no variables that moderated placebo response across all disorders besides diagnostic indication. There were also no significant predictors when examining anxiety and OCD trials specifically. However, among depressive disorders, more study sites, more study visits and fewer number of subjects per site and fewer subjects getting medication per site was associated with a higher placebo response.
Meta-Regression Analysis of Predictors of Placebo Response
The table displays results of meta-regression of trial level and subject level predictors of placebo response with values of PE, upper and lower CI and p-value. Statistically significant results (p < 0.05) are in bold.
CI, confidence intervals; OCD, obsessive-compulsive disorder; PE, point estimates; SSRI, selective serotonin receptor inhibitor.
Discussion
This meta-analysis set out to investigate potential trial level and subject level predictors of placebo symptom improvement and placebo response both within and across diagnoses. We demonstrated that response and symptom improvement in placebo groups were heterogeneous and influenced by certain trial and sample characteristics. In line with previous pediatric research, we found that both outcomes were highest in depression compared with anxiety disorders and OCD (Bridge et al. 2009). Table 4 shows how symptom improvement and response in placebo groups were predicted by trial and subject level characteristics.
Direction of Correlation of Significant Predictors with Placebo Outcome Measures
The table provides the direction of correlations of all the statistically significant predictors of placebo symptom improvement and placebo response.
↑ Positive correlation.
↓ Negative correlation.
— Nonsignificant.
OCD, obsessive-compulsive disorder.
The most striking finding is that both the magnitude and predictors of placebo response in depression did not replicate across anxiety and OCD. This could be owing to either the intrinsic nature of depression—its specific pathophysiology (Cohen et al. 2008)—or a result of how we measure it or contexts specific to pediatric depression trials (Walkup 2017). For instance, depression is considered a “disease of hopelessness”; thus, patients with depression may be more responsive to “hope” in the form of a placebo than those with anxiety or OCD. Thus, certain symptoms unique to depression may be more amenable to the influence of a placebo. In addition, because of high comorbidity between depression, anxiety, and OCD, it is plausible that some of the placebo improvement seen in anxiety and OCD could be owing to the placebo alleviating depression. A Research Domain Criteria (RDoC) approach could aid in understanding the placebo response without the constraints of diagnostic categories and examine the particular symptoms likely to improve with placebo. Alternatively, because these trials do not utilize a no-treatment arm, it cannot be ruled out that the differences observed in magnitude of placebo response is because of differences in the natural history of the three disorders rather than the placebo itself. In addition, we cannot distinguish between placebo effects and nonspecific benefits of clinical trial participation, which may be present in the placebo group of these trials. With regard to predictors specifically, it is possible that it is easier to see their influence in depression because it has a larger placebo response. In addition, the depression trials were the greatest in number increasing statistical power.
Trial characteristics
Across all disorders a greater number of study sites were associated with higher placebo symptom improvement. However, when analyzed according to the indication, depression trials appear to be responsible for contributing toward these results—as neither anxiety nor OCD trials shows placebo response and symptom improvement to be significantly predicted by this variable. In fact, apart from OCD showing lower placebo improvement in trials conducted in the United States, trial design characteristics were shown to significantly moderate only the depression trials.
One key reason for greater number of study sites were associated with greater placebo symptom improvement could be the regulatory context in which most of the recent pediatric depression trials were conducted as discussed by Walkup (2017). The Food and Drug Administration (FDA) was concerned about the practice of prescribing antidepressant to children despite the dearth of RCTs showing efficacy or safety within that population. To address this, the FDA offered pharmaceutical industries a 6-month patent extension if they conducted pediatric trials of the drug for the same indication as was used in adults. It was stipulated that these trials had to be published before the expiry of the patent. For drugs like Prozac, in exchange for a relatively modest investment in clinical studies, the patent extension would yield a significant $500 million in additional profit (“Eli Lilly: Life After Prozac” 2001). Given the large financial incentives awarded toward completing these trials within the FDA mandated timeline, often speed of trial completion was prioritized over design decisions to maximize signal detection. Multiple sites including nonacademic sites were used to satisfy the required participant quota. In fact, depressive trials tended to have higher sample sizes because they are powered to detect smaller effect sizes as higher placebo improvement is anticipated in these trials. In addition, trials that have the pressure to fulfill a larger subject recruitment quota, quickly and efficiently, could tend toward lower quality control in subject selection and ratings. This leads to an increased assessment variance and the risk of loss of sensitivity to detect the efficacy signal.
Quality control and consistency of procedure are especially challenging when there are geographically separate and heterogeneous sites. Thus, in line with previous research, an increased number of sites was associated with a greater placebo response across disorders and when adjusting for indication. This is because the greater the number of sites, the more challenging it is to run a trial correctly, train researchers well, and have inter-rater reliability. In addition, trials with a greater number of sites may have lower standards (e.g., experience with pediatric patients and with the underlying disorder) for the inclusion of particular sites and researchers in the trial. A higher number of subjects recruited per site was associated with a lower placebo response in both analyses. This is not surprising as a higher number of subjects per site is a result of fewer sites. This suggests that as sites have more experience running subjects in trials that the placebo response may decrease. This result calls into question the current trend toward a greater number of sites per trial, which could be one of the reasons for the increase of the placebo response over time.
Consistent with previous studies, later publication year was associated with greater placebo symptom improvement (Cohen et al. 2010). There are several explanations of why later year of publication shows a positive correlation with placebo improvement. First, to conduct large-scale studies to prove efficacy within a short period of time, there has been a trend toward using multiple study sites. Keck et al. (2000) found that inter-rater reliability and differences in population were potential problems that influenced placebo effect. Second, later studies are more likely to recruit participants who have previously been on similar antidepressant medication and thus expectancy causes them to respond similarly to the placebo. Finally, there is a greater time lag between the publication of failed trials, which may be because of higher placebo effect, as opposed to successful trials (Reyes et al. 2011). Thus, studies not actually conducted but only published later would demonstrate a higher placebo effect. We were unable to find reliable data on recruitment dates for many of the included trials so were unable to include this as a predictor of interest.
In addition, presence of a placebo lead-in period did not substantially effect magnitude of placebo symptom improvement in pediatric antidepressant trials. This result is consistent with previous research in adult antidepressant studies (Trivedi and Rush 1994). There could be several reasons for this. First, subjects may receive verbal or social cues from the unblinded study staff. Faria et al. (2017) compared two single-blind placebo lead-in depression trials with one that had a double-blind placebo lead-in. They found that the double blind identified 28% of the participants as placebo responders, in comparison with the 10% identified by the single blind. Furthermore, the placebo lead-in results in increased trial duration. Increased trial duration is associated with greater placebo response (Rutherford et al. 2009). Conversely, Gelenberg et al. (2008) have argued that perhaps longer placebo lead-in periods are needed to distinguish the placebo responders. However, there are ethical implications of increasing the duration of studies that give a placebo to a clinical pediatric population. These may be sidestepped by an alternative method of providing an initial course of psychotherapy and selecting the nonresponders for the next phase of the clinical trial. This makes the assumptions that those who do not have a therapeutic response to the psychosocial contexts of therapy are unlikely to respond to a placebo pill.
Subject-level characteristics
Several subject-level factors such as age and baseline measures have been reported in previous studies to moderate placebo response in adults and children (Rutherford and Roose 2013). Overall we found little evidence that age and other subject-level characteristics were associated with magnitude of placebo response. That being said, we were limited to examining aggregate level study characteristics associated with magnitude of placebo response. It remains quite possible that subject-level factors at an individual level are associated with placebo response such as age, psychiatric comorbidity, socioeconomic status, or race/ethnicity that we either could not examine or had limited power to examine with study-level data.
Limitations
There are several limitations of our study that are worth mentioning. First, none of the included RCTs compared placebo response across disorders; hence, only indirect comparisons can be made. Trials used different rating scales to measure symptoms that decrease the consistency of effect sizes across trials. However, we controlled for this to a certain extent by using SMD as opposed to weighted mean difference. It should be noted however, that using SMD can lead to a reduction of information and granularity. In addition, because of lack of individual patient data, we ran a meta-analysis of aggregate subject data across trials that has reduced statistical power. Absence of individual level data precluded us from examining other potentially important moderators of placebo response such as psychiatric comorbidity. Current research suggests there is a high degree of overlap of comorbidity between depression, anxiety disorders, and OCD. However, using aggregate data to assess degree of psychiatric comorbidity in samples can be quite problematic as there is substantial inconsistency across trials in how secondary diagnoses are defined, measured, and reported.
We excluded the trials that lacked data without contacting the authors of the trials and did not include unpublished data in our meta-analysis. This led to a limited number of trials within disorders and increased chance of publication bias. Because we only had a limited number of trials, this decreased power for between-groups analysis.
It is important to note the limitations of meta-analysis as a method of research. Although meta-analysis provides more precise and a better estimate of the relationship that exists in the population than single studies, the results might be limited by the selection of an incomplete set of studies, inclusion of studies that lack internal construct, external construct and statistical conclusion validity, presence of studies with small sample sizes, and heterogeneity of methods used in studies. Thus, it should be emphasized that a meta-analysis is only as strong as the included studies in the analysis. If the included studies are a biased sample of all possible studies, then the mean effect calculated by meta-analysis will reflect this bias. We cannot exclude the possibility of any type of bias in the included studies.
Conclusion
In conclusion, our results found that because the characteristics of placebo response do not replicate across disorders, they cannot be extrapolated from one disorder to the next. Thus, to minimize the placebo response, the specific disorder should be considered. Based on our results, across disorders, minimizing the number of study sites would significantly reduce placebo improvement. In addition to these, we could specifically decrease the placebo response in depression trials by increasing the number of subjects per site and subjects on study medication, minimizing the number of study visits and conducting the studies in the United States. Further research is needed into the predictors of the pediatric placebo response in anxiety and OCD.
Clinical Significance
Depressive disorder, anxiety disorders and OCD are among the most prevalent psychopathologies in children and adolescents. They are disabling and are associated with adverse long-term outcomes; therefore, early diagnosis and treatment are critical. Placebo response has been identified as an important factor influencing the success of adult antidepressant trials, yet little research of placebo response has been conducted in pediatric populations. Utilizing the predictors of pediatric placebo response, we found could aid in designing successful child psychopharmacological trials so that medications can reach the clinical setting.
Key Points
What's known: Magnitude of placebo response is a key factor in determining the success of psychotropic medication trials. However, very little is known about it in the pediatric population.
What's new: Apart from number of study sites and public funding—the predictors of placebo response do not replicate across internalizing disorders in children. Increased placebo response in depression trials was associated with decreased number of subjects per site, increased number of study visits, and conducting studies outside the United States. Further research is needed on predictors of placebo response in anxiety and OCD RCTs.
What's relevant: Identifying potential predictors specific to the pediatric placebo response is crucial in the development of pediatric psychopharmacology. In addition, better understanding contributors to placebo response might allow clinicians to maximize the placebo effect in the therapeutic setting and/or minimize the need for unnecessary pharmacological treatments.
Footnotes
Disclosures
M.H.B. receives research support from Therapix Biosciences, Neurocrine Biosciences, Janssen Pharmaceuticals, and Biohaven Pharmaceuticals. M.H.B. gratefully acknowledges additional research support from NIH, NARSAD, and the Patterson Foundation.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.