
Abstract
Objective:
The goal of this study was to evaluate the efficacy and safety of duloxetine in children and adolescents (9–17 years of age) with major depressive disorder (MDD) in Japan.
Methods:
This study consists of two clinical trials. First, a 6-week, randomized double-blind placebo-controlled clinical trial (RCT) was conducted. The primary endpoint of RCT was the change in Children's Depression Rating Scale-Revised (CDRS-R) total scores from baseline. Following RCT, an open-label long-term extension trial (OLE) was conducted to investigate the longer-term safety of duloxetine for ∼1 year.
Results:
In RCT, CDRS-R total score changes from baseline to 6 weeks after the start of administration (primary endpoint) were −21.03 in the duloxetine group (n = 74) and −22.42 in the placebo group (n = 74). No significant difference was observed in the primary endpoint between the groups (p = 0.5587). In addition, no significant difference was observed in secondary endpoints such as CDRS-R response rates. The proportion of patients with ≥1 treatment-emergent adverse event (TEAE) in RCT was significantly higher in the duloxetine group (78.7%) than in the placebo group (62.2%), and most were mild or moderate in severity. Changes in CDRS-R total scores during OLE, in consecutive patients from the duloxetine group in RCT (n = 63), or placebo group (n = 59) in RCT, and newly enrolled patients (n = 28), were −12.1, −11.3, and −17.8, respectively. The proportion of patients with ≥1 TEAE in OLE was 90.5%, 88.1%, and 89.3% in the respective groups, and most of them were mild or moderate in severity.
Conclusions:
Duloxetine did not show superiority to placebo in efficacy in children and adolescents with MDD in Japan. Overall reported TEAEs were consistent with the currently available duloxetine safety profile and no new safety finding was observed in the two clinical trials.
Introduction
Major depressive disorder (MDD) is a common illness worldwide, and different from the usual mood fluctuations and short-lived emotional responses to challenges in everyday life. Especially, when long-lasting with moderate or severe intensity, untreated MDD may become a serious health condition. It can cause the affected person to suffer greatly, and function poorly at work, at school, and in the family (World Health Organization, 2020a). MDD can occur in children and adolescents, and it is diagnosed using the same Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) diagnostic criteria as is used in adults.
However, as noted in DSM-5, irritable mood can be used instead of depressive mood in children and adolescents with MDD (American Psychiatric Association 2013). Epidemiological studies have reported that the first episodes of MDD can appear at an early age, the frequency rises rapidly at about 10–12 years, and the incidence rate in adolescents is not significantly different from that reported in adults (Zisook et al. 2007). The 1-year prevalence rate of depression has been estimated to be 2.8% in children younger than 13 years, and 5.6% in adolescents in Western counties (Costello et al. 2006; Thapar et al. 2012), compared to 4.4% in adults (World Health Organization 2017). Similarly, the estimated prevalence rate is 1% and 5.6% in children and adolescents in Japan (Doi et al., 2001; Denda 2006).
In children and adolescents, MDD is reported to be a leading cause of disease burden (World Health Organization 2020b). There are also reports of its negative impact on family and friends (Lynch and Clark 2006; Bhatia and Bhatia 2007). With regard to treatment, there are two broad therapeutic approaches consisting of psychosocial interventions and pharmacological therapy. The World Health Organization highlights the benefits of psychotherapy and recommends psychosocial treatment for patients with mild disease (World Health Organization, 2020a,c) and a similar conclusion was reported in a recent network meta-analysis (Liang et al. 2021).
Pharmacological therapy with antidepressants is generally reserved for more severe cases of child and adolescent depression or in situations when psychotherapy has failed to work (Shain and the Committee on Adolescence 2016; Luxton and Kyriakopoulos 2021). However, to date, when it comes to drug therapy, only fluoxetine for pediatric patients 8–18 years of age (Emslie et al. 1997) and escitalopram for adolescents 12–17 years of age (Emslie et al. 2009) have been approved by the U.S. Food and Drug Administration (FDA). In Japan, no medication has been approved for child and adolescent depression. Furthermore, there is little published evidence relating to pharmacological treatment for child and adolescent patients with depression, and no randomized double-blind placebo-controlled clinical trial (RCT) has been completed.
Duloxetine is a serotonin-noradrenaline reuptake inhibitor, which has proven to be effective in adults with MDD worldwide, including in Japanese patients (Detke et al. 2002; Goldstein et al., 2002; Higuchi et al. 2009; Cipriani et al. 2012). In addition, this medication has been approved by the FDA for generalized anxiety disorder and fibromyalgia in pediatric patients. With regard to MDD, two Phase 3 clinical trials evaluated duloxetine in children and adolescents with MDD in the United States and other countries, but the results were inconclusive because neither duloxetine nor a positive control (fluoxetine) was shown to be more effective than placebo (Atkinson et al. 2014; Emslie et al. 2014).
The continued detrimental impact of child and adolescent depression suggests that there is an unmet need for early and better diagnostic approaches and treatment options. We therefore conducted a 6-week RCT and an open-label long-term extension trial (OLE) to evaluate the efficacy and safety of duloxetine in child and adolescent patients with MDD in Japan.
Methods
The following two clinical trials were conducted. An RCT for 6 weeks in child and adolescent patients with MDD was conducted in 36 medical institutions in Japan between December 2017 and November 2019 (
The study protocols and informed consent forms were approved by the Institutional Review Boards at each study site. The study was performed in accordance with Good Clinical Practice guidelines and in line with the principles set out in the Declaration of Helsinki.
Study design
RCT consisted of four periods (Fig. 1A). First, a screening period during which patient consent was obtained and subjects considered suitable at Visit 1 was provisionally enrolled into the clinical trial and monitored for 1–3 weeks. During the treatment period, eligibility was confirmed at Visit 2 and selected patients were registered, and randomly assigned (1:1) to receive duloxetine or placebo orally once daily after breakfast for 6 weeks in a double-blinded manner. Duloxetine dosage was started at 20 mg/day, and increased to 40 mg/day after 1 week on Visit 3. One week later (Visit 4 onward), if the Clinical Global Impressions-Severity (CGI-S) score was 3 or higher without any safety concern, the dose level was increased to 60 mg/day. If any safety issue occurred after increasing the dosage to 60 mg/day, then a dosage reduction to 40 mg/day was allowed. When the CGI-S score was 3 or higher, and the safety issue resolved, a dose increase to 60 mg/day was allowed again on Visit 6 or Visit 7. At the completion of treatment (Visit 8) or at the time of early discontinuation, a 1-week tapering period was initiated to minimize possible adverse events (AEs) resulting from the discontinuation of the study drug. If the dosage level in the duloxetine or placebo groups was equivalent to 60 mg/day at the time of discontinuation or completion of the treatment period, then the study drug was reduced to 40 mg/day for the first 3 days, followed by 20 mg/day for the last four days. If the dose level was 40 mg/day, the study drug was administered at 20 mg/day throughout the 1-week tapering period. Finally, a 1-week follow-up period was conducted to evaluate possible AEs, which might develop after the study drug was discontinued.

Study schedule:
OLE included the patients requesting to continue treatment from RCT as well as newly enrolled patients (NEW). OLE also comprised four periods (Fig. 1B). For consecutive patients, patient consent was obtained until end of observation at Visit 9 in RCT and eligibility was confirmed at Visit L2 and selected patients were registered to receive treatment with duloxetine once daily after breakfast for 50 weeks. For NEW, patient consent was obtained during the screening period and eligible patients were provisionally enrolled into the clinical trial (Visit L1) and monitored for 1–3 weeks. Eligibility was confirmed at Visit L2 and selected patients were registered to receive treatment with duloxetine.
Dosage was started at 20 mg/day and increased to 40 mg/day after 1 week (Visit L3). If there was no safety concern, the dosage was increased to 60 mg/day at least 2 weeks after starting treatment. Subsequently, duloxetine dosage could be increased/decreased (between 40 and 60 mg/day) depending on symptoms as necessary. At the completion of treatment (Visit L17) or at the time of early discontinuation, a 1–2 week tapering period was initiated to minimize possible AEs resulting from the discontinuation of the study drug. If the dose level was 60 mg/day at this time, then the duloxetine dosage was reduced to 40 mg/day for 1 week followed by 20 mg/day for 1 week. If the dose level was 40 mg/day, the dosage was reduced to 20 mg/day for 1 week. Finally, a 1-week follow-up period was conducted to evaluate possible AEs, which might develop after the study drug was discontinued.
Psychoeducation
Four video sessions of psychoeducation were conducted in all patients in RCT and in new patients in OLE. The contents were “What is depression?,” “Drug therapy and course of symptoms,” “Need for rest and continued drug therapy,” and “Course of symptoms and prevention of recurrences.” The patients in RCT viewed them at visits 1, 2, 5, and 8 (or at the time of early discontinuation) and the new patients in OLE at visits L1, L2, L5, and L6 (or at the time of early discontinuation), respectively.
Patients
Male or female patients between 9 and 17 years of age were eligible if they were diagnosed with MDD or persistent depressive disorder with major depressive episode, based on definitions of the DSM-5 (American Psychiatric Association 2013), and had a first episode at the age of 7 years or older. The Mini-International Neuropsychiatric Interview for Children and Adolescents (MINI-KID version 7.0.2) was used for diagnosis of depression (Sheehan et al. 2010). Investigators were trained in the use of MINI-KID. In RCT, patients with a Children's Depression Rating Scale-Revised (CDRS-R) (Poznanski et al. 1984) total score of 40 or higher and a CGI-S (Guy 1976) of 4 or higher at Visits 1 and 2 were enrolled.
The legally authorized representative ensured that the patient observed all trial procedures, and they also signed the voluntary consent form before the patient's participation in the clinical trial. For patients ≥13 years of age, informed assent in writing was also obtained and, wherever possible, it was also procured from younger patients (9–13 years of age).
Key exclusion criteria for RCT were as follows: patients with ≥30% improvement of the CDRS-R total score from Visit 1 to Visit 2; complications or medical history, including neurodevelopmental disorders, schizophrenia or other psychotic disorders, bipolar/related disorders, and trauma- or stress-related disorders, disruptive, impulse-control, and conduct disorders (DSM-5); suicidal ideation and suicide attempt within 1 year before Visit 1; and a patient answering “Yes” in the Columbia-Suicide Severity Rating Scale (C-SSRS) suicidal ideation question 4 (Active suicidal ideation with some intent to act, without specific plan), question 5 (Active suicidal ideation with specific plan and intent), or the questions about the suicidal behavior, except for nonsuicidal self-injurious behavior at Visits 1 and 2.
For OLE, both new patients and consecutive patients from the preceding RCT were eligible if they met the entry criteria. Briefly, new male or female patients were eligible if they had a diagnosis of depressive illness, were between 9 and 17 years of age, and had a first episode at 7 years of age or older. Key inclusion criteria were MDD (as defined for RCT) with no criteria for CDRS-R total and CGI-S scores. Exclusion criteria included suicidal ideation and suicide attempt within 1 year before Visit 1 and complications or medical history such as neurodevelopmental disorders, schizophrenia or other psychotic disorders, bipolar/related disorders, and trauma- or stressor-related disorders, disruptive, impulse-control, and conduct disorders (DSM-5) similar to RCT.
Efficacy evaluation
The clinician-rated CDRS-R total score based on the sum of all 17 items (Poznanski et al. 1984) and the 7 grades of the CGI-S (Guy 1976) efficacy measures were collected at every visit by the investigator/subinvestigator. Investigators were trained in the use of the CDRS-R rating scale by watching relevant case videos.
In RCT, the primary endpoint was the change in CDRS-R total score from baseline to week 6. Secondary endpoints included the percentage of patients who had a decrease in the CDRS-R total score of 30% or more/50% or more compared with the baseline value (30% response/50% response rate, respectively). It was planned to calculate response rates using raw baseline and postbaseline scores, but response rates were also calculated and evaluated after subtracting the minimum 17 points from raw scores (Emslie et al. 2002, 2014; Atkinson et al. 2014). Additional secondary endpoints were the subscales of CDRS-R and CDRS-R item 13 (suicidal ideation), and the change in CGI-S score from baseline to week 6. Remission was defined as a CDRS-R total score less than 28 (Poznanski and Mokros 1996).
In OLE, efficacy was assessed by the CDRS-R total, CDRS-R item 13 (suicidal ideation), and CGI-S scores.
Safety evaluation
AEs were monitored throughout the clinical trials. Safety was assessed in terms of AEs, which were classified by MedDRA (version 20.1) system organ classes and preferred terms. Among the AEs reported in the Case Report Forms, those which occurred after the first study drug administration and ongoing were classed as treatment-emergent adverse events (TEAEs) for the safety analysis. In OLE, an event that occurred in RCT and remained during continued long-term treatment was also considered to be a TEAE.
Statistical methods
The target sample size for RCT was 148 patients (74/group), which was calculated to detect an effect size of 0.49 with power of ≥80% based upon a two-sample t-test at a two-sided significance level of 0.05, and a dropout rate of 10%.
Efficacy analyses were performed in the full analysis set (FAS), which consisted of all randomized patients who received the study drug at least once, and with a CDRS-R total score measured at baseline and another one measured after study drug administration. A mixed-effects model for repeated measures (MMRM) approach was used for the primary efficacy analysis. A linear model with unstructured covariance was applied, using change in the CDRS-R total score from baseline as the response variable; treatment group, time point, and interaction between the treatment group and time point as fixed effects; and the total CDRS-R score at baseline and age (<12 years, ≥12 years) as covariates. The Kenward–Roger approximation was used to adjust the degree of freedom. Secondary efficacy endpoints were analyzed as follows: 30% (or 50%) response rate and remission rate; the number of responders and the response rate were calculated by treatment group. A Cochran–Mantel–Haenszel test with age as stratification factor was used to compare the duloxetine and placebo groups. CDRS-R subscale and Item 13 (suicidal ideation) and CGI-S were also analyzed using the MMRM approach in a similar manner to the primary efficacy analysis.
Safety analyses were performed for all randomized patients who received at least one dose of the study drug. The numbers and proportions of patients with TEAEs, death, serious AEs (SAEs), suicide-related TEAEs, and TEAEs leading to discontinuation of the treatment were tubulated for each treatment group. Suicide-related TEAEs were defined as TEAEs that belong to the Standardised MedDRA Queries (version 20.1) of suicide/self-injury.
In OLE, the target sample size was to enroll 135 patients with the goal of achieving 100 patients complete the 50-week treatment period. Efficacy and safety data were summarized descriptively for consecutive patients from the duloxetine or placebo groups in RCT, as well as NEW.
Statistical tests were performed at a two-sided significance level of 0.05 and no multiplicity adjustment was made. All analyses were performed using SAS Version 9.4.
Results
Randomized double-blind placebo-controlled clinical trial
A total of 149 patients were enrolled and randomized to treatment; 75 in the duloxetine group and 74 in the placebo group (Fig. 2). One patient in the duloxetine group did not have any observation and was excluded from the FAS.
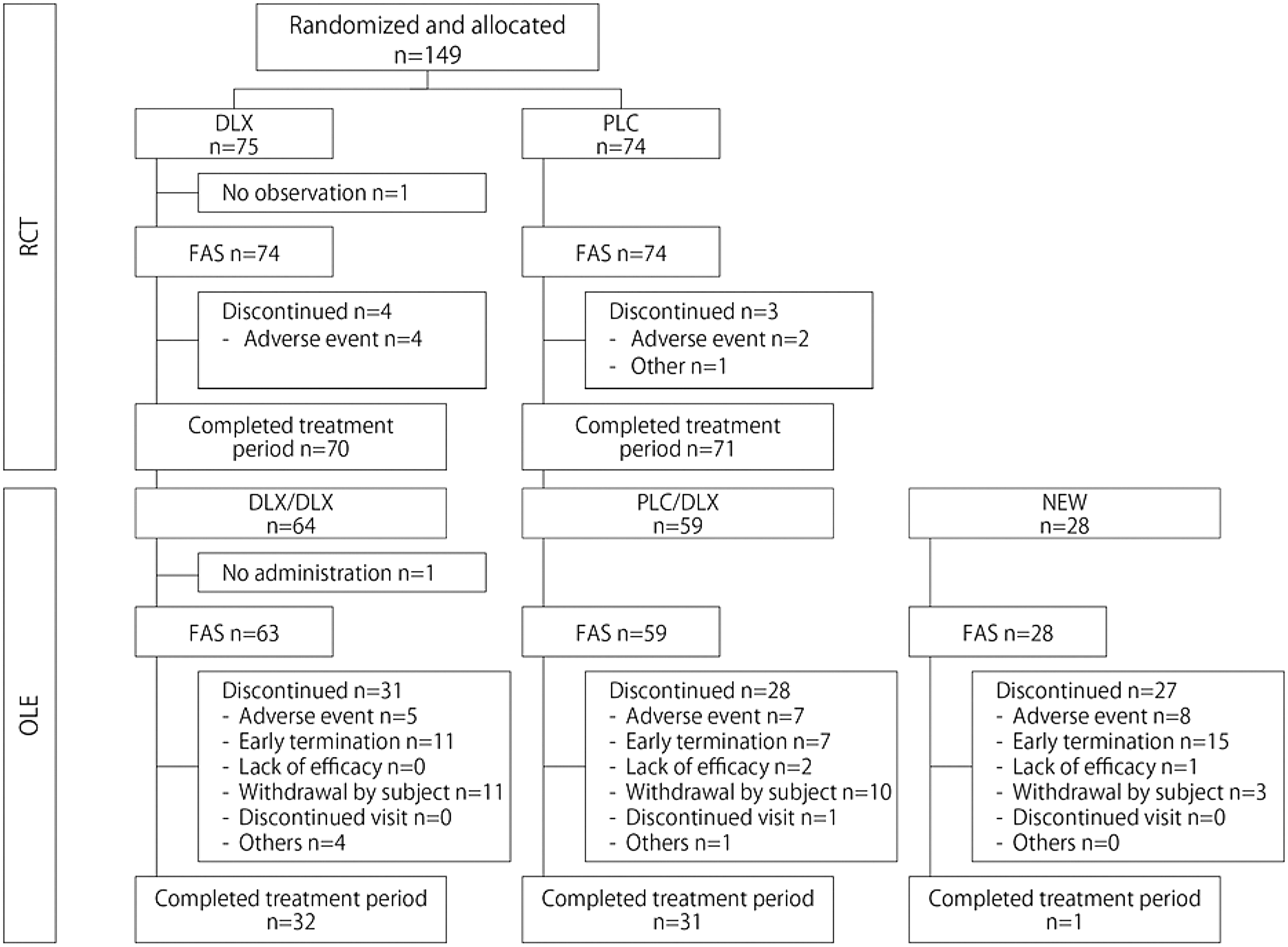
Patient disposition in RCT and OLE. DLX, duloxetine; DLX/DLX, consecutive patients from the duloxetine group in RCT; FAS, full analysis set; NEW, newly enrolled patients; OLE, open-label long-term extension trial; PLC, placebo; PLC/DLX, consecutive patients from the placebo group in RCT; RCT, randomized double-blind placebo-controlled clinical trial.
The baseline characteristics and demographics of the two randomized groups were balanced (Table 1). Complications present in at least 10% patients in the RCT were seasonal allergy, headache, rhinitis allergic, and dysmenorrhea in the duloxetine group and seasonal allergy, headache, rhinitis allergic, dysmenorrhea, and dermatitis atopic in the placebo group. As for anxiety, there were no patients with social anxiety disorder in the duloxetine group and 3 of 74 (4.1%) in the placebo group.
Patient Demographics for the Full Analysis Set of RCT and OLE
Baseline score at the initiation of respective trial.
Baseline score at the initiation of RCT.
CDRS-R, Children's Depression Rating Scale-Revised; CGI-S, Clinical Global Impressions-Severity; DLX, duloxetine; DLX/DLX, consecutive patients from the duloxetine group in RCT; DSM-5, Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; MDD, major depressive disorder; NEW, newly enrolled patients; OLE, open-label long-term extension trial; PLC, placebo; PLC/DLX, consecutive patients from the placebo group in RCT; RCT, randomized double-blind placebo-controlled clinical trial; SD, standard deviation.
The baseline values for CDRS-R total score were similar between the duloxetine (mean ± standard deviation [SD]; 65.7 ± 10.5, n = 74) and the placebo groups (mean ± SD; 63.3 ± 12.4, n = 74) (Table 1). The proportions of patients receiving duloxetine at final doses of 40 and 60 mg were 27.0% (20/74 patients) and 73.0% (54/74 patients), respectively.
The least square (LS) mean (±standard error [SE]) change from baseline to week 6 in CDRS-R total score during the course of RCT (primary endpoint) was −21.03 ± 2.04 in the duloxetine group and −22.42 ± 2.05 in the placebo group. There was no significant difference between duloxetine and placebo (LS mean difference [95% confidence interval {CI}]: 1.39 [−3.30 to 6.08]; p = 0.5587; Fig. 3). As shown in Table 1, patients younger than 12 years accounted for 6.8% and 6.8% in duloxetine and placebo groups, respectively. The mean change from baseline to last observation on the CDRS-R total score in patients younger than 12 years was −12.51 ± 5.29 (LS mean ± SE) and −27.70 ± 5.30 in duloxetine and placebo groups, respectively. For those 12 years of age and older, it was −19.03 ± 1.75 and −19.38 ± 1.75, respectively. There were no statistically significant differences between duloxetine and placebo for the change in CDRS-R total scores within either age group (LS mean difference [95% CI]: 15.20 [−5.34 to 35.74], p = 0.1103; 0.36 [−4.54 to 5.26], p = 0.8850). Duloxetine and placebo showed similar improvements from baseline also in secondary endpoints, including CDRS-R 30% response rate, 50% response rate, remission rate, changes from baselines in CDRS-R subscales (mood, somatic, subjective, and behavior), CDRS-R item 13 (suicidal ideation), and CGI-S score (Table 2). As described in Methods, we conducted two ways for calculating the response rates, which were (a) calculation with raw scores and (b) calculation with values after subtracting the minimum 17 points from raw scores.

LS mean change from baseline in CDRS-R total score in RCT (Full Analysis Set, MMRM). CDRS-R, Children's Depression Rating Scale-Revised; DLX, duloxetine; LS mean, least square mean; MMRM, mixed-effects model for repeated measures; PLC, placebo; RCT, randomized double-blind placebo-controlled clinical trial; SE, standard error.
Secondary Endpoints in the Full Analysis Set of RCT
Last observation carried forward.
Calculated with raw baseline and postbaseline scores.
Calculated with baseline and postbaseline values after subtracting the minimum 17 points from raw scores.
CDRS-R, Children's Depression Rating Scale-Revised; CGI-S, Clinical Global Impressions-Severity; CI, confidence interval; DLX, duloxetine; LS, least square; PLC, placebo; RCT, randomized double-blind placebo-controlled clinical trial; SE, standard error.
The 30% response rates were (a) 48.6%/(b) 59.5% in the duloxetine group and (a) 43.2%/(b) 59.5% in the placebo group. The 50% response rates were (a) 21.6%/(b) 32.4% and (a) 21.6%/(b) 32.4% in duloxetine and placebo groups, respectively. For RCT, CDRS-R item-13 (suicidal ideation) baseline scores were 1.9 ± 1.1 in the duloxetine group and 1.8 ± 1.0 in the placebo group (mean ± SD). The change from baseline in CDRS-R item 13 (suicidal ideation) was −0.65 ± 0.10 for duloxetine and −0.62 ± 0.10 for placebo (LS mean change ± SE), and the difference was not statistically significant (−0.04 [−0.25 to 0.18] LS mean difference [95% CI], p = 0.7409).
The proportion of patients reporting at least one TEAE was significantly higher in the duloxetine group [153 events in 59 of 75 patients (78.7%)] than in the placebo group [86 events in 46 of 74 patients (62.2%)] (p = 0.0319, Fisher's exact test). No deaths and no serious TEAE occurred. All TEAEs were mild or moderate in severity and the most common TEAEs in the duloxetine group (n = 75) were nausea (25.3%), somnolence (22.7%), decreased appetite (17.3%), and nasopharyngitis (13.3%) (Table 3). Suicide-related TEAEs were observed in 5 of 75 patients in the duloxetine group and 2 of 74 patients in the placebo group (Table 4). The individual TEAEs were suicidal ideation in 2 patients, suicide attempt in 1 patient, and intentional self-injury in 3 patients in the duloxetine group, and suicidal ideation in 1 patient, suicide attempt in 1 patient, and intentional self-injury in 1 patient in the placebo group. The investigators considered that TEAEs in 2 of 5 patients (4 of 8 events) in the duloxetine group and 1 of 2 patients (1 of 3 events) in the placebo group were unrelated to the study drugs.
Treatment-Emergent Adverse Events Occurring With Placebo or Duloxetine (in at Least 10% of Patients in One of the Groups): Descending Order Based on Consecutive from Placebo Group in RCT and OLE
DLX, duloxetine; DLX/DLX; consecutive patients from the duloxetine group in RCT; NEW, newly enrolled patients; OLE, open-label long-term extension trial; PLC, placebo; PLC/DLX, consecutive patients from the placebo group in RCT; RCT, randomized double-blind placebo-controlled clinical trial; TEAE, treatment-emergent adverse event.
Suicide-Related Treatment-Emergent Adverse Events in RCT and OLE
Suicide-related TEAEs were defined as TEAEs that belong to the Standardised MedDRA Queries (version 20.1) of suicide/self-injury.
DLX, duloxetine; DLX/DLX, consecutive patients from the duloxetine group in RCT; NEW, newly enrolled patients; OLE, open-label long-term extension trial; PLC, placebo; PLC/DLX, consecutive patients from the placebo group in RCT; RCT, randomized double-blind placebo-controlled clinical trial; TEAE, treatment-emergent adverse event.
TEAEs leading to discontinuation of treatment occurred in 4 patients in the duloxetine group (suicidal ideation, suicide attempt, decreased appetite, and aggression in 1 event each; and nausea in 2 events) and 2 patients in the placebo group (akathisia, suicidal ideation, and suicide attempt in 1 event each).
Open-label long-term extension trial
A total of 151 patients were enrolled, 64 consecutive patients from the duloxetine group in RCT (DLX/DLX group), 59 consecutive patients from the placebo group in RCT (PLC/DLX group), and 28 NEW group (Fig. 2). One patient in the DLX/DLX group met exclusion criteria for alanine aminotransferase and did not receive any study drug. Thus, this patient was excluded from the FAS and safety analysis sets, and 150 patients were treated with duloxetine (Table 1). Once the results of RCT were analyzed and showed no significant advantage for duloxetine over placebo, the sponsor terminated the ongoing OLE early, and this led to withdrawal of 33 patients. Consequently, the median duration for treatment was 302.5 days, and 103 patients were treated for >24 weeks and 64 patients completed the 50-week treatment period in OLE. The proportions of patients receiving duloxetine at a maximum dose of 40 or 60 mg were 23.3% and 74.0%, respectively.
CDRS-R total scores for the DLX/DLX group, PLC/DLX group, and NEW group over the duration of RCT and OLE periods are individually shown in Figure 4. The DLX/DLX group and PLC/DLX group already had improved CDRS-R total scores at the baseline of OLE. In OLE, CDRS-R total scores gradually improved from baseline values in all the groups. The change from baseline to last observation (mean ± SD) was −12.1 ± 15.4 (from 45.8 ± 16.0 to 33.8 ± 13.6) in the DLX/DLX group (n = 63), −11.3 ± 16.3 (from 43.6 ± 14.4 to 32.2 ± 16.8) in the PLC/DLX group (n = 59), and −17.8 ± 19.7 (from 59.1 ± 13.4 to 41.4 ± 20.1) in the NEW group (n = 28).

Mean CDRS-R total scores in the RCT and OLE. CDRS-R, Children's Depression Rating Scale-Revised; DLX/DLX, consecutive patients from the duloxetine group in RCT; LOCF, last observation carried forward; NEW, newly enrolled patients; OLE, open-label long-term extension trial; PLC/DLX, consecutive patients from the placebo group in RCT; RCT, randomized double-blind placebo-controlled clinical trial; SD, standard deviation.
Likewise, CGI-S scores declined, that is, the changes from baseline to last observation (mean ± SD) were −0.8 ± 1.2 (from 3.6 ± 1.0 to 2.8 ± 1.0) in the DLX/DLX group (n = 63), −0.8 ± 1.3 (from 3.5 ± 0.9 to 2.7 ± 1.3) in the PLC/DLX group (n = 59), and −1.3 ± 1.4 (from 4.6 ± 0.9 to 3.4 ± 1.4) in the NEW group (n = 28). The changes from baseline to last observation in CDRS-R item-13 (suicidal ideation) (mean ± SD) were −0.1 ± 0.6 (from 1.3 ± 0.6 to 1.2 ± 0.6) in the DLX/DLX group, −0.1 ± 0.8 (from 1.3 ± 0.7 to 1.3 ± 0.8) in the PLC/DLX group, and −0.4 ± 1.1 (from 1.8 ± 1.0 to 1.5 ± 1.1) in the NEW group.
Regarding safety, 57 of 63 in the DLX/DLX group (90.5%), 52 of 59 in the PLC/DLX group (88.1%), and 25 of 28 in the NEW group (89.3%) reported TEAEs. The most common TEAEs were nausea, somnolence, and nasopharyngitis (Table 3). All were rated as mild or moderate in severity. There were no AEs whose frequency increased with long-term administration.
Suicide-related events are reported in Table 4. The number of patients with ≥1 suicide-related TEAE was 6 of 63 in the DLX/DLX group (9.5%), 10 of 59 in the PLC/DLX group (16.9%), and 6 of 28 in the NEW group (21.4%). Intentional self-injury was responsible for the majority of suicide-related TEAEs. The investigators considered that 18 of 23 events were unrelated to the study drug.
No death occurred during OLE treatment. SAEs were observed in three patients and these were mania in the DLX/DLX group, abnormal behavior in the PLC/DLX group, and suicide attempt in the NEW group. The suicide attempt was judged to be possibly related to the study drug by the investigator and the others were not. All SAEs were moderate in severity, with recovery or improvement confirmed.
TEAEs leading to study discontinuation of the treatment occurred in 20 patients (5 patients in the DLX/DLX group; diarrhea, intentional self-injury, liver function test increased, mania, somnolence, and suicidal ideation, each in 1 patient; 7 patients in the PLC/DLX group; intentional self-injury in 3 patients, somnolence in 2 patients, and abnormal behavior, irritability, orthostatic hypotension, and thirst, each in 1 patient; and 8 patients in the NEW group; nausea in 4 patients, suicidal ideation and/or attempt in 3 patients, somnolence in 2 patients, decreased appetite, hallucinations mixed, intentional self-injury, and malaise, each in 1 patient).
Discussion
To our knowledge, this RCT is the first completed RCT to evaluate the efficacy and safety of antidepressant therapy in children and adolescents with MDD in Japan. However, no significant difference was shown for duloxetine compared with placebo, with respect to efficacy endpoints. We conducted a subgroup analysis by stratifying by age 12 years, but there were no statistically significant differences between duloxetine and placebo in either subgroup. This is similar to previous studies on duloxetine, where no significant differences were shown in either subgroup (Atkinson et al. 2014; Emslie et al. 2014). However, the proportion of patients younger than 12 years in this study was only 7%, which was much less than about 40% in previous studies.
As a consequence of the primary results, OLE was terminated early, but it should be noted that CDRS-R total scores gradually improved during the long-term treatment. As for the safety profile, the AEs in clinical trials are already well known for duloxetine and no new safety signals were identified during RCT and OLE.
A majority of previous studies involving children and adolescents with depression have reported a failure to identify significant benefit with active treatment over placebo (Cohen et al. 2008; Tsapakis et al. 2008). An increase in placebo response overtime in RCTs of antidepressants for pediatric depression has previously been reported (Cohen et al. 2008) and it has been hypothesized that younger patients may be more responsive to placebo than adults (Parellada et al. 2012; Weimer et al. 2013; Meister et al. 2020). Therefore, the inability to detect differences between duloxetine and placebo in this RCT may also be attributed to large placebo responses. There have been relatively few positive studies concerning antidepressant therapy in children and adolescents with MDD.
Those that have been reported include two clinical trials with fluoxetine in children and adolescents, and another with escitalopram in adolescents (Emslie et al. 1997, 2002, 2009). CDRS-R total scores for placebo at baseline/endpoint were 57.6/47.1 in a fluoxetine study reported in 1997 (Emslie et al. 1997) and 55.1/40.2 in another fluoxetine study reported in 2002 (Emslie et al. 2002). The changes in CDRS-R were estimated to be −10.5 and −14.9 by simple subtraction, respectively. In the escitalopram study, the change from baseline to end of treatment was −18.8 for placebo (Emslie et al. 2009). In this study, the change in baseline/endpoint was −22.42, indicating a relatively larger placebo response, and no statistically significant superiority of duloxetine relative to placebo was observed.
There are several possible reasons for the higher placebo response observed in our study. First, psychoeducation might have contributed to the higher placebo response. Second, patients and parents were informed about the well-established efficacy of duloxetine in previous clinical trials involving adult patients with MDD when the purpose of the clinical trial was explained to them to obtain informed assent and informed consent. This might have created an expectation of high efficacy in the placebo group as well.
In this clinical trial, psychoeducation was uniformly conducted by watching the same preprepared educational video, instead of the participating medical institutions conducting their own psychoeducation. Psychoeducation is important in the treatment of pediatric depression; however, the methodology used may vary between clinical sites and this might impact individual responses to treatment. Therefore, we conducted uniform psychoeducation across all sites aiming to minimize variability in clinical effects and enhance detection power. In addition, pediatric patients and parents may be worried about participation in clinical trials, especially those that are placebo controlled. We took the view that psychoeducation would help reduce any concern/anxiety by providing a clearer understanding about depression and thought that this would assist patient inclusion and retention in these clinical trials. However, rather than diminish the variability, the psychoeducation potentially might have provided some positive benefit to patients in both the duloxetine and placebo groups (through better communication and addressing some of the concerns of the patients/family). Such a positive effect has been reported in adolescents with depression (Bevan Jones et al. 2018) and might have masked the efficacy of duloxetine.
The safety profile of duloxetine in these clinical trials was similar to that reported for Japanese adults (Higuchi 2009; Higuchi et al. 2009), as well as in children and adolescents treated in non-Japanese clinical trials (Atkinson et al. 2014; Emslie et al. 2014). The most common TEAEs in duloxetine-treated patients were nausea, somnolence, and nasopharyngitis and there were no AEs whose frequency increased with long-term administration. The overall safety data reported in this study are considered to be consistent with the well-documented safety profile of duloxetine currently available.
The FDA issued a black-box warning of suicide risk for inclusion in the package inserts for all antidepressants used in the treatment of children, adolescents, and young adults in 2004 (U.S. Food and Drug Administration 2004, Farnaro et al. 2019). This highlights that the suicide risk associated with antidepressants used to treat pediatric patients with MDD and other psychiatric disorders is a particularly important consideration. Similar precautions have been applied to Japanese package inserts. In recent years, observational research and reanalysis of clinical studies have suggested that the risks of worsening symptoms and ultimately suicide by not prescribing antidepressants to adolescents are higher than the suicide risks of prescribing them (Gibbons et al. 2007, 2012).
In the clinical trials in this study, suicide-related TEAEs were reported as 11 events in 7 patients in RCT [5/75 patients (6.7%) in the duloxetine group and 2/74 (2.7%) in the placebo group], and 28 events in 22 patients in OLE [6/63 (9.5%) in DLX/DLX group; 10/59 (16.9%) in PLC/DLX group; and 6/28 (21.4%) in NEW group]. The incidence of intentional self-injury accounted for majority of the suicide-related TEAEs and appears to be numerically higher in the duloxetine group than in the placebo group in RCT. The investigators considered that most cases of intentional self-injuries were unrelated to duloxetine (4 of 5 events in the duloxetine group in RCT and 18 of 23 events in OLE). Most of the events without causal relation were due to temporary stress or anxiety resulting from life events (4 of 4 events in the duloxetine group in RCT and 15 of 18 events in OLE). Consistently, the change of CDRS-R item-13 (suicidal ideation) did not show an increase in suicide risk in either the RCT or OLE. It is reported that nonsuicidal self-injury appears during adolescence, particularly during the mid-years (15–17 years) and declines in early to mid-adulthood (Plener et al. 2015). In young people, self-harm behavior is often an expression of distress and desire for escape from troubling situations (Hawton and James 2005).
Limitations
RCT period (6 week) might be relatively short because CDRS-R total scores continued decreasing in the long term (up to additional 50 weeks). The early termination of OLE did not allow us to observe all patients for the full duration of the planned treatment period, so it might be insufficient to provide the information than expected from long-term therapy.
Conclusions
Duloxetine did not show superior efficacy compared with placebo, and reported AEs across RCT and OLE were consistent with its currently known profile and no new safety finding or concern was identified.
Clinical Significance
This is the first completed RCT to evaluate the efficacy and safety of antidepressant therapy in children and adolescents with MDD in Japan.
Footnotes
Authors' Contributions
All authors contributed to the article and approved this submitted version.
Acknowledgments
The authors would like to thank all patients, investigators, and staff who participated in the clinical trials, and Dr Steve Clissold (Content Ed Net, Japan) for editorial support, which was funded by Eli Lilly Japan K.K., Kobe, Japan, and Shionogi & Co., Ltd., Osaka, Japan.
Disclosures
M.I., A.N., and T.O. are employees of Shionogi & Co., Ltd., Osaka, Japan. H.K. is an employee Eli Lilly Japan, Kobe, Japan, and a shareholder of Eli Lilly & Co., Indianapolis, USA. T.S. received research grants from Shionogi & Co., Ltd., Takeda Pharmaceutical, consulting fees from Mochida Pharmaceutical, Viatris, Taisho Pharmaceutical, Sumitomo Dainippon Pharma, personal fees from Shionogi & Co., Ltd., Takeda Pharmaceutical, Sumitomo Dainippon Pharma, Taisho Pharmaceutical, Chugai Pharmaceutical, Jansen Pharmaceutical, and Otsuka Pharmaceutical, Nobelpharma, support for attending meetings from Sumitomo Dainippon Pharma, Taisho Pharmaceutical, and received the endowed chair by City of Sapporo, Hokkaido, Japan. H.M. reports receiving grant form Shionogi & Co., Ltd. and honoraria from Shionogi & Co., Ltd., and Eli Lilly Japan. All other authors declare no conflicts of interest.