
Abstract
Objective:
Evaluate the long-term improvement and safety of aripiprazole in treating irritability in Asian children and adolescents (6–17 years) with autistic disorder.
Methods:
A 52-week, open-label, flexibly dosed (2–15 mg/day) study on the improvement and safety of aripiprazole in patients with autistic disorder who had completed an antecedent 12-week open-label study. The evaluation of efficacy was conducted using the Aberrant Behavior Checklist (ABC), Clinical Global Impression (CGI) scale, Child Yale-Brown Obsessive-Compulsive Scale (CY-BOCS), Vineland Adaptive Behavior Scale (VABS), and the Parenting Stress Index-Short Form (PSI-SF). Safety and tolerability measurements included adverse events, vital signs, electrocardiography, laboratory tests, body weight, and extrapyramidal symptoms (EPSs).
Results:
During the 52-week treatment, all effectiveness variables, including ABC, CGI, CY-BOCS, VABS, and PSI-SF scores, showed improvement. Regarding safety, the proportion of patients who experienced any treatment-emergent adverse events (TEAEs) was 58.62% (34/58 subjects, 75 cases). The most common TEAE was nasopharyngitis reported in 20.69% (15/58 subjects, 15 cases) and the other TEAE with an incidence of ≥10% was weight increases in 18.97% (11/58 subjects, 11 cases). Of them, 27.59% (16/58 subjects, 28 cases) experienced adverse drug reactions (ADRs). The most common ADR was weight increase reported in 15.52% (9/58 subjects, nine cases). The incidence of serious adverse events (SAEs) was 5.17% (3/58 subjects, three cases), which were epiphysiolysis, seizure, and a suicide attempt, but these were not ADRs. There were no clinically significant changes found in the evaluation of EPSs.
Conclusions:
Aripiprazole showed improvement for behavioral problems and adaptive functioning and was well tolerated in patients with autistic disorder until nearly a year after drug use.
The Clinical Trial Registration number: NCT 02069977
Introduction
Autistic disorder is a neurodevelopmental disorder with childhood onset characterized by deficits in social communication and social interaction and the presence of restricted, repetitive patterns of behavior, interest, or activity. In addition to core symptoms, subjects with autistic disorders frequently exhibit irritability such as aggression, tantrums, and self-harm behavior, disrupting their school and family environment (McGuire et al. 2016).
For treating such troublesome irritability, atypical antipsychotics have become widely used in patients with autistic disorders (Schubart et al. 2014). However, these drugs can cause metabolic adverse effects in children and adolescents, especially weight gain (Almandil et al. 2013). Risperidone and aripiprazole are the only Food and Drug Administration (FDA)-approved medications for treating irritability associated with autistic disorder. Risperidone is effective for treating irritability associated with autistic disorder (DeFilippis and Wagner 2016). Still, increased appetite, weight gain, and enuresis are associated with long-term risperidone use (Aman et al. 2015), and hyperprolactinemia can occur (Almandil et al. 2013).
Aripiprazole, one of the other FDA-approved drugs for irritability with autistic disorder, carries a lower risk of extrapyramidal symptoms (EPS), sedation, and hyperprolactinemia (Fung et al. 2016). Also, it is believed to have a more favorable metabolic profile than other atypical antipsychotics (Douglas-Hall et al. 2011). It is a relatively new drug with a unique mechanism of action as a potent partial agonist of dopamine D2 receptors, in addition to properties as a 5-HT1A agonist and 5-HT2 antagonist, different from other antipsychotics (Burris et al. 2002). Aripiprazole is postulated to work through the mechanism of functional selectivity, where the D2 functional effects are dependent upon the cellular location of the D2 receptor (Shapiro et al. 2003). The functional selectivity hypothesis proposes that whether an agent is an agonist, partial agonist, or antagonist depends upon the cellular milieu.
Alterations in dopaminergic and serotonergic neurotransmission have been implicated in autistic disorder, with abnormalities in these systems demonstrated through neuroimaging and metabolic studies (Posey et al. 2008). As aripiprazole has an affinity for both dopamine and serotonin receptors, it is likely to exert its action through similar mechanisms as other antipsychotic medications. Its 5-HT2 antagonism is higher compared with most atypical antipsychotics, but lower compared with ziprasidone or risperidone. However, its degree of antagonist activity, along with its partial D2 agonist activity, is at an optimal level, reducing the risks of extrapyramidal signs such as tardive dyskinesia. It also has moderate H1 receptor affinity, which decreases the risk of sedation and weight gain compared to clozapine and olanzapine, which have high affinities for the H1 receptor (Owen et al. 2009).
Several previous studies have been conducted on the efficacy and safety of aripiprazole. However, most of them were randomized controlled trials, insufficient for generalization to real-world clinical settings. Also, most of them have the limitation of a short-term follow-up period (Marcus et al. 2009; Owen et al. 2009; Robb et al. 2011; Ichikawa et al. 2017). Another 52-week open-label study was limited to a Western population (Marcus et al. 2011a). Although the FDA approved aripiprazole to treat irritability in patients with autism in November 2009 with supporting evidence, there is a lack of data on Asian populations.
To the best of our knowledge, only one long-term study has investigated the efficacy and safety of aripiprazole in an Asian population, but it was limited to Japanese patients (Ichikawa et al. 2018). In this context, it was thought to be valuable to perform this study, to assess the long-term improvement and safety of aripiprazole involving patients with an autistic disorder from multinational countries in Asia.
Methods
The study was a multinational, multicenter, 52-week open-label, single-arm design study. Children and adolescents with autistic disorder were recruited from eight hospitals, four hospitals in South Korea, three in the Philippines, and one in Thailand. The Institutional Review Boards of the participating institutions approved this study. All parents/guardians provided written informed consent, and subjects provided written informed assent when possible.
Study design
The study design is shown in Figure 1. All psychotropic medications (including antipsychotic, antidepressant, anxiolytic, mood-stabilizing, and neuroleptic agents) were washed out during the screening period and should be prohibited during the study. Medications that may influence prolactin were also prohibited during the study. Risperidone and aripiprazole should be discontinued for at least 1 and 3 weeks, respectively. However, the medications for attention-deficit/hyperactivity disorder (ADHD), such as methylphenidate, atomoxetine, and clonidine, were allowed to continue without washout periods if the drug had been administered for ≥12 weeks at a stable dose for ≥4 weeks (Kim et al. 2018). However, a dose increase of ADHD medications was not permitted during the study.
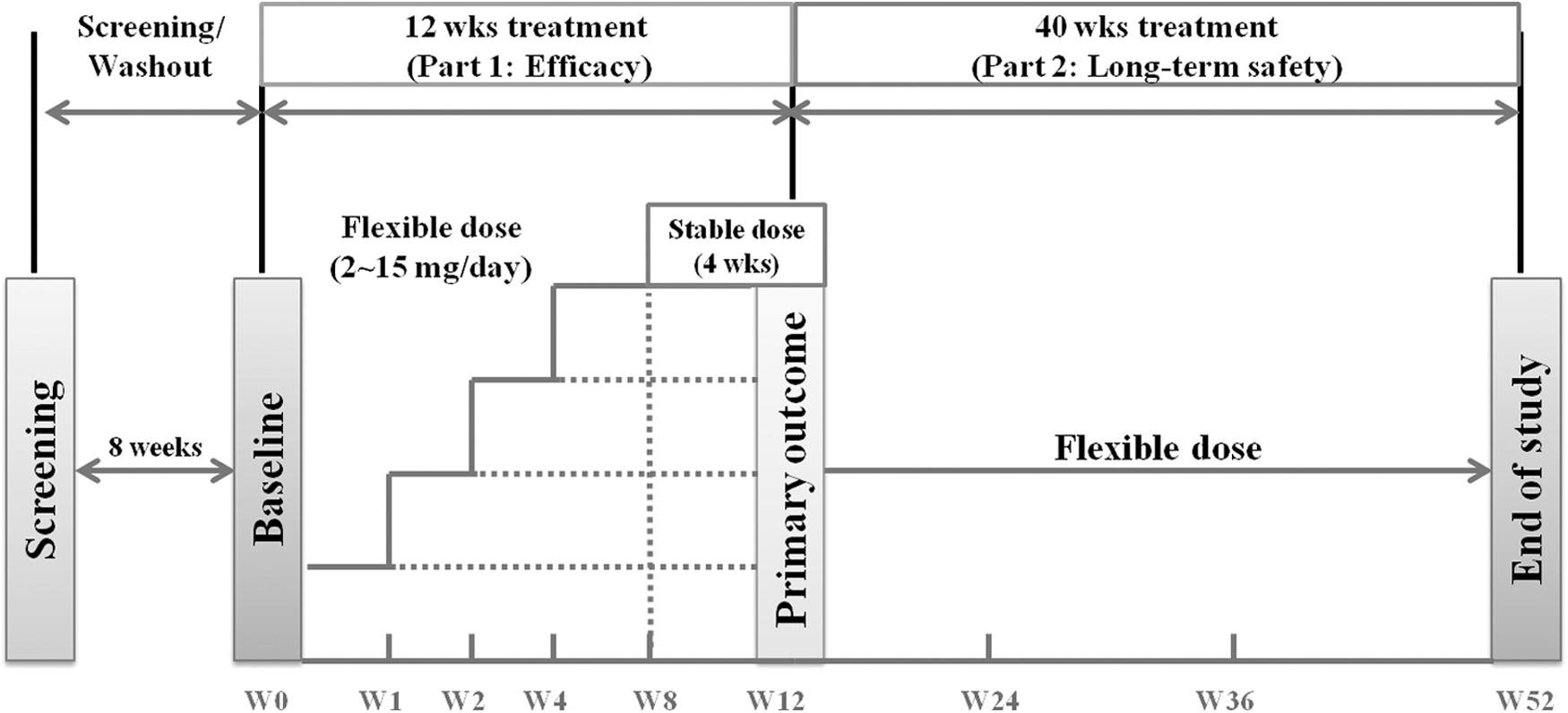
Study design: multinational, multicenter, 52-week open-label single-arm study.
Any medication for a concomitant therapy was permitted if it is not defined as the prohibited medications in the study protocol. Lorazepam or alprazolam, for anxiety related to medical/study procedures, and sleep aids (diphenhydramine, or nonbenzodiazepine hypnotics such as zolpidem and zaleplon) could be administered at the investigator's discretion. Benztropine or propranolol based on investigator judgment was permitted for the treatment of EPSs, but not within 12 hours before administration of movement rating scales.
Subjects who met all other eligibility criteria at the baseline visit began the 12-week dose adjustment phase (Part 1). Subjects were guided to visit the clinic at the end of treatment on weeks 1, 2, 4, 8, and 12, at the time when samples for effectiveness and safety measurements were to be collected. The subjects who completed the 12-week treatment were eligible for the long-term phase study (Part 2) to evaluate the safety and tolerability of aripiprazole.
The treatment was continued when clinically warranted as judged by the investigator, and no clinically relevant adverse events (AEs) would preclude inclusion in the study. The patients visited the clinic at weeks 24, 36, and 52 when samples for effectiveness and safety measurements were to be collected.
The result of the 12-week, Part 1 study has already been reported (Kim et al. 2018). This study aims to evaluate the long-term improvement and safety of aripiprazole up to 52 weeks from the baseline based on data from the Part 2, extension phase.
Study patients
The inclusion criteria were (1) 6–17 years of age; (2) diagnosis of autistic disorder defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) (American Psychiatric Association 2000) and confirmed using the Autism Diagnostic Interview-Revised (ADI-R); (3) behavioral problems such as irritability, agitation, and/or self-injurious behavior; (4) a Clinical Global Impression Severity of Illness scale (CGI-S) score of ≥4 and an Aberrant Behavior Checklist-Irritability (ABC-I) subscale score of ≥18 at screening and baseline; and (5) mental age of ≥24 months (Kim et al. 2018).
The exclusion criteria were (1) Rett's disorder, childhood disintegrative disorder, Asperger's disorder, or pervasive developmental disorder not otherwise specified according to the DSM-IV-TR; (2) schizophrenia, other psychosis, and mood disorders, including bipolar disorder and major depressive disorder according to the DSM-IV-TR criteria; (3) significant risk of committing suicide based on the subject's medical history or a routine mental status examination; (4) seizure in the past year; (5) history of severe head trauma or stroke; (6) history of neuroleptic malignant syndrome; (7) resistance to antipsychotic medication; and (8) the presence of a significant comorbid medical illness (Kim et al. 2018).
Comorbid psychiatric disorders, including intellectual disability and ADHD, were included and diagnosed based on the DSM-IV-TR. The intelligence quotient was measured by one of the standardized methods (Kim et al. 2018).
Dosing
The initial dose of aripiprazole was 2 mg/day and it was allowed to increase based on the response up to the target dosage of 5–15 mg/day. Dose increases were done gradually by titration and were scheduled by investigators weekly or for each study visit. During the Part 1 study, dose decreases were allowed at the investigator's discretion at any time due to tolerability. However, no adjustment was allowed after week 8 (Visit 6) to keep the level of treatment stable for the final 4 weeks of treatment. During Part 2, dose adjustments could be made at any time based on the efficacy and tolerability of the current dose. The mean daily dose of aripiprazole across the entire 52 weeks was 6.0 ± 3.1 mg (range 2–15 mg).
Assessments
To confirm the diagnosis of autistic disorder, we used solely the ADI-R (Lord et al. 1994) conducted by skilled interviewers who had been previously trained and approved as “research reliable” on ADI-R or who had completed a 2-day rater training course conducted by a certified ADI-R trainer.
The effectiveness and safety assessments were performed at baseline, weeks 1, 2, 4, 8, 12, 24, 36, and 52, and at the time of early termination when applicable. The primary outcome measure was the mean change in the caregiver-rated ABC-I subscale (Aman et al. 1985) scores from baseline to week 12.
The secondary outcome measures included clinician-rated mean changes in CGI-S scores, other Aberrant Behavior Checklist (ABC) subscale scores (lethargy/social withdrawal, stereotypy, hyperactivity, and inappropriate speech), scores on the Clinical Global Impression Improvement of Illness scale (CGI-I), the Child Yale-Brown Obsessive-Compulsive Scale (CY-BOCS) (Scahill et al. 1997), the Vineland Adaptive Behavior Scale (VABS) (Sparrow et al. 2005), and the Parenting Stress Index-Short Form (PSI-SF) (Abidin 1995). The ABC, CGI-S, and CGI-I were assessed at baseline and weeks 1, 2, 4, 8, 12, 24, 36, and 52. The CY-BOCS, VABS, and PSI-SF were assessed at baseline, and weeks 12 and 52.
Safety assessments, including treatment-emergent adverse event (TEAE), adverse drug reaction (ADR), serious adverse event (SAE), vital signs, and body weight, were conducted at each visit. A TEAE is any untoward medical occurrence in a patient administered an investigational product (IP) that does not necessarily have a causal relationship with this drug. An ADR is a special type of AE in which a causative relationship can be shown. SAE, based on patient outcome criteria, is usually associated with events that threaten a patient's life or functioning. The presence and severity of EPSs were also assessed at each visit using the Simpson-Angus Scale (SAS), the Abnormal Involuntary Movement Scale, and the Barnes Akathisia Rating Scale. Twelve-lead electrocardiography and laboratory tests were performed at baseline, week 12, and at the end of treatment.
Statistical analyses
The safety sample included all subjects who received at least one dose of the study medication. The effectiveness sample included all subjects in the safety sample who completed at least one follow-up effectiveness evaluation and had a corresponding baseline value. The primary outcome measure, the difference in ABC-I scores between baseline and week 12, was analyzed using a Wilcoxon signed-rank test. For secondary outcome measures and safety assessments, the paired t-test or Wilcoxon's signed-rank test with Bonferroni's correction for multiple comparisons (p < 0.05/number of comparisons) was used. All statistical analyses were performed using a two-sided test and at a 5% level of significance. The last-observation-carried-forward imputation was used for subjects who did not complete the treatment period. All statistical analyses were performed using SAS software (version 9.2; SAS Institute, Inc.) (Kim et al. 2018).
Results
All 67 eligible subjects enrolled in the study received the IP and were included in the Part 1 safety set. Among those, eight subjects were prematurely withdrawn due to protocol deviations, the withdrawal of informed consent, and a lack of efficacy. Thus, the remaining 59 subjects completed Part 1. No subject was withdrawn from the study due to AEs.
Afterward, 58 subjects were included in the Part 2 safety set, excluding 1 subject who did not join the Part 2 study. Among those, 8 subjects were prematurely withdrawn, and the remaining 50 subjects completed the Part 2 study. Among 58 subjects, 54 subjects who completed at least 1 follow-up assessment during Part 2 were included in the effectiveness sample. Subject disposition is shown in Figure 2.
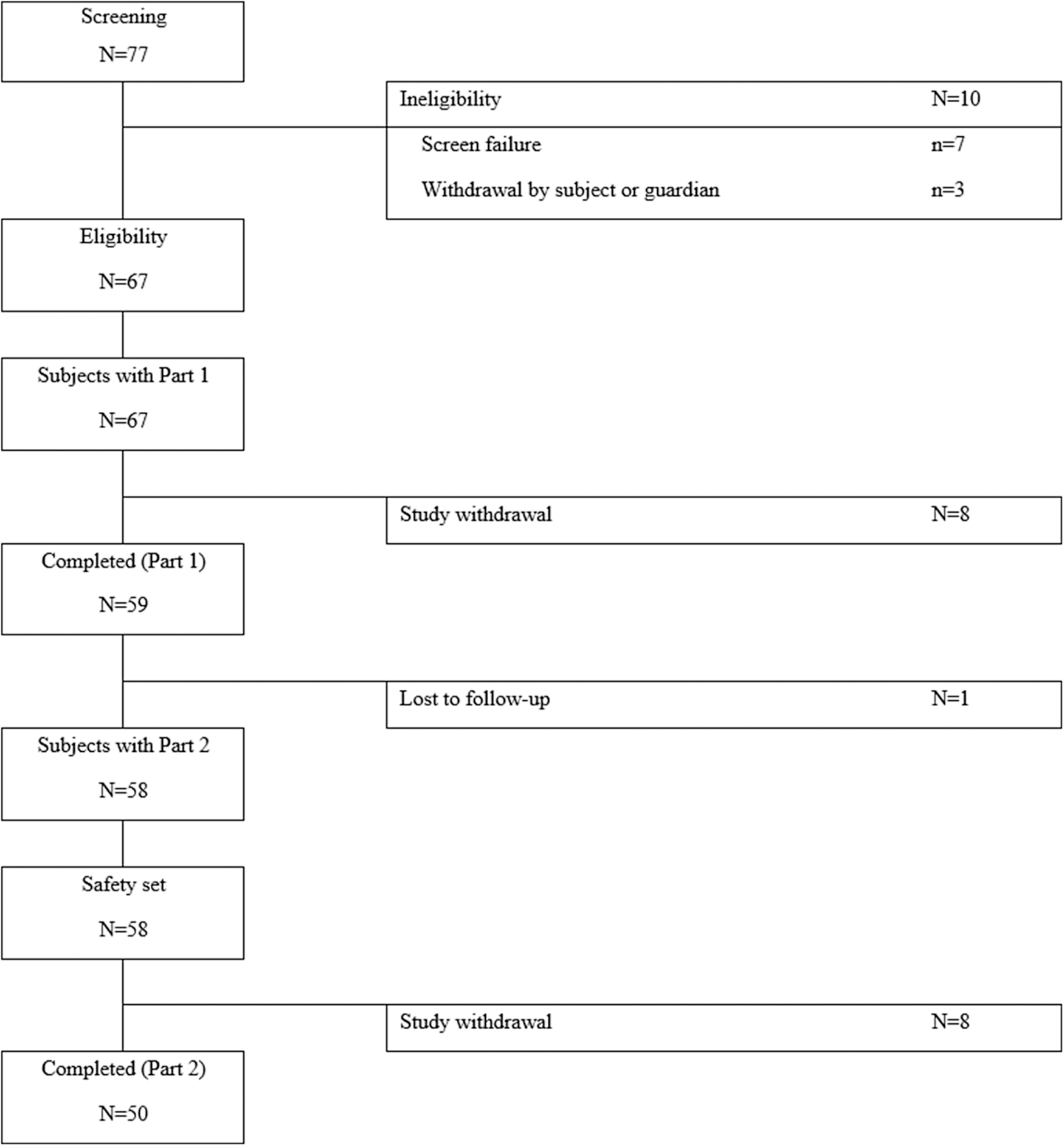
Subject disposition.
Demographic and other baseline characteristics
The demographic and clinical characteristics of the subjects in Part 2 are shown in Table 1. The mean subject age was 10.0 ± 3.1 years, and most subjects were boys (n = 44, 81.5%) and younger than 12 years (n = 39, 72.3%). The mean weight and height were 40.3 ± 13.9 kg and 140.9 ± 16.6 cm, respectively, resulting in a mean body mass index (BMI) of 19.71 ± 4.02 kg/m2. The mother, father, and grandparents were the main caregivers for 77.78%, 16.67%, and 5.56% of the subjects, respectively.
Part 2 Demographic and Clinical characteristics
Overlap count.
ADHD, attention-deficit/hyperactivity disorder; SD, standard deviation.
In our study, 17 subjects changed from risperidone and 9 subjects previously received aripiprazole. Nine subjects maintained their ADHD medication. Except for ADHD medication, the proportion of subjects who administered any concomitant medication during the study period was 53.70% (29/54 subjects, 282 cases). When classified by therapeutic subgroup for further details, the most frequently administered class was cough and cold preparations in 18.52% (10/54 subjects, 69 cases), followed by antihistamines for systemic use and nasal preparations in 12.96% each (7/54 subjects, 56 and 11 cases each).
Effectiveness
The effectiveness variables during the 12-week treatment, including the mean ABC-I score change, were discussed in our previous study (Kim et al. 2018). The results of secondary outcome measures during the Part 2 study are shown in Table 2. During the 52-week treatment, ABC-I scores showed a statistically significant improvement from baseline to every time point before week 52 (all p < 0.0001), and the other subscales for lethargy, stereotypy, hyperactivity, and inappropriate speech also showed a statistically significant improvement from baseline to every time point of weeks 1–52 (all p < 0.05). The improvements in the ABC subscale scores are also illustrated in Figure 3.
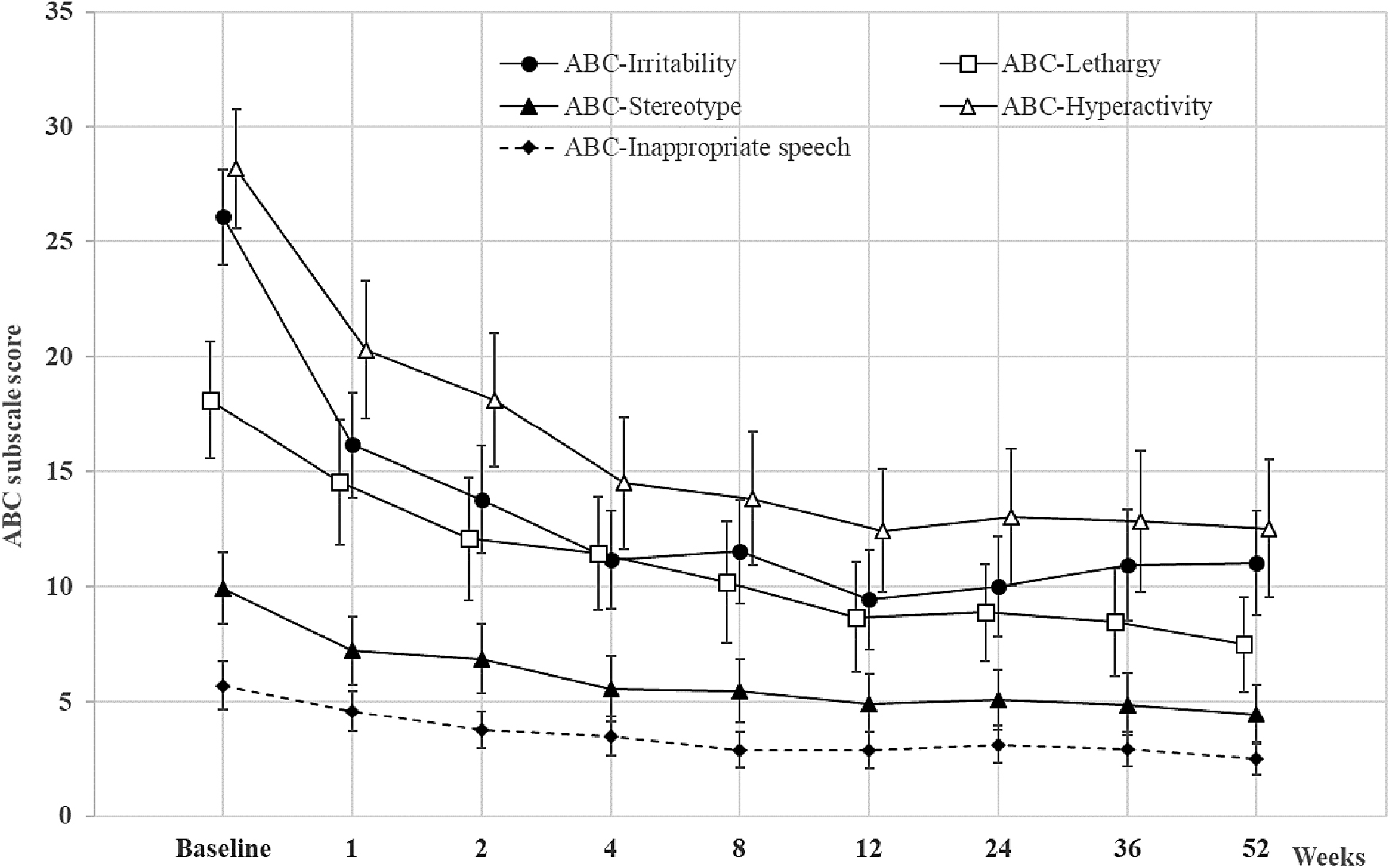
Mean change from baseline in the ABC subscale scores (LOCF, efficacy sample). Data are expressed as mean with CI. ABC, aberrant behavior checklist; CI, confidence interval; LOCF, last observation carried forward.
Results of Secondary Outcome Measures (Last Observation Carried Forward, Effectiveness Sample, N = 54)
Multiple-comparison adjusted significance level: p < 0.01.
Number of sample = 26.
CI, confidence interval.
The severity assessed using CGI-S showed statistically significant decreases, which indicated improvement in severity from baseline to every time point (all p < 0.05). The mean score for improvement (CGI-I), a 7-point scale (from 6 Much worse to 1 Very much improved) that requires the clinician to assess how much the patient's illness has improved or worsened relative to a baseline state, also showed a decreasing trend over time, indicating the degree of improvement.
The mean obsession and compulsion subtotal scores and the total CY-BOCS scores showed statistically significant decreases from baseline to weeks 12 and 52, which indicated an improvement in the severity of the obsessive-compulsive behaviors (all p < 0.05).
The results of the VABS for communication, daily living skills, and socialization showed statistically significant increases from baseline to weeks 12 and 52 (all p < 0.05). The mean VABS score for maladaptive behavior showed statistically significant decreases from baseline to weeks 12 and 52 (all p < 0.0001), but no statistically significant change was observed in motor skill scores (all p > 0.05). The overall adaptive behavior composite VABS score showed statistically significant increases at each time point (week 12, p = 0.0004; week 52, p = 0.0149).
The total stress score assessed using the PSI also showed a statistically significant decrease of 13.4 ± 18.8 and 13.9 ± 18.0 from baseline to weeks 12 and 52, respectively, indicating improvement in stress (p < 0.0001).
Adverse events
In the safety set, three subjects (3/58 subjects, 5.17%) experienced a total of three AEs before IP administration, and the proportion of patients who experienced any TEAE was 94.83% (55/58 subjects, 237 cases). Of those, the TEAE incidence rate was 84.48% (49/58 subjects, 162 cases) in Part 1 and 58.62% (34/58 subjects, 75 cases) in Part 2.
Safety variables during the 12-week treatment in Part 1 were discussed in our previous study (Kim et al. 2018). For Part 1, the most common TEAE was weight increases reported in 25.86% (15/58 subjects, 15 cases), and the other TEAEs with an incidence ≥of 10% were somnolence and sedation in 20.69% each (12/58 subjects, 16 and 14 cases, respectively), nasopharyngitis in 18.97% (11/58 subjects, 14 cases), and pyrexia in 13.79% (8/58 subjects, eight cases). Of them, 67.24% (39/58 subjects, 108 cases) of the subjects experienced ADRs. The most common ADR was weight increase reported in 25.86% (15/58 subjects, 15 cases), and the other ADRs with an incidence ≥10% were sedation in 20.69% (12/58 subjects, 14 cases) and somnolence in 18.97% (11/58 subjects, 15 cases). A SAE, tonsillitis, was reported in one patient (1/58 subjects, 1.72%), but it was not an ADR.
For Part 2, during the 52-week treatment, the proportion of patients who experienced any TEAEs was 58.62% (34/58 subjects, 75 cases). The most common TEAE was nasopharyngitis reported in 20.69% (12/58 subjects, 15 cases) and the other TEAEs with an incidence ≥10% was weight increases in 18.97% (11/58 subjects, 11 cases). The TEAEs of the subjects are presented in Table 3.
Part 2 Treatment-Emergent Adverse Events of Subjects
Of them, 27.59% (16/58 subjects, 28 cases) experienced ADRs. The most common ADR was weight increases reported in 15.52% (9/58 subjects, nine cases). The incidence of SAEs was 5.17% (3/58 subjects, three cases) and included epiphysiolysis, seizure, and a suicide attempt (1/58 subjects, 1.72%, 1 case, respectively), but the SAEs were not ADRs.
Bodyweight and metabolic abnormalities
The proportion of subjects with weight gain in the Part 1 study was 25.86% (15/58 subjects) and the proportion in the Part 2 study was 18.97% (11/58 subjects). The mean BMI change during the 52-week aripiprazole treatment was 1.4 ± 1.7 kg/m2 (Table 4).
Mean Change of Body Mass Index During the 52-Week Aripiprazole Treatment
LOCF, last observation carried forward; OC, observed case; SD, standard deviation.
Five clinically significant laboratory results were seen in two subjects in the safety set during the study period. One subject had an alanine aminotransferase level of 78, 56, and 148 U/L (normal range 0–40 U/L) and an aspartate aminotransferase level of 52, 42, and 83 U/L (0–40 U/L) at baseline, week 12, and week 52, respectively, representing abnormal results above the upper limit of normal. In the case of the other subject, the triglyceride levels were 320.37, 371.70, and 478.79 mg/dL (normal range, <149.57 mg/dL) at baseline, week 12, and week 52, respectively, indicating clinically significant abnormal results.
Moreover, the total cholesterol levels were 205.79, 220.46, and 208.88 mg/dL (normal range <200.77 mg/dL) at baseline, week 12, and week 52, respectively, and the fasting blood glucose levels were 90.10, 102.89, and 107.58 mg/dL (normal range at ages 6–11 years, 60–100 mg/dL) at baseline, week 12, and week 52, respectively, showing abnormal results above the upper limit of normal.
Prolactin
Aripiprazole treatment was associated with a decrease in serum prolactin. At baseline, the mean serum prolactin was 9.32 ± 7.93 ng/mL. The serum prolactin levels showed statistically significant decreases of 7.69 ± 7.85 and 5.45 ± 11.36 ng/mL from baseline to weeks 12 and 52, respectively (p < 0.0001).
Other safety measurements
No clinically significant change was found in the vital sign evaluations. The PR interval, although an electrocardiogram (EKG) result, showed a statistically significant change (increased) (p < 0.0023), but there was no clinical significance. Also, no clinically significant change was found in the evaluation of EPSs.
Discussion
This long-term open-label study demonstrated that flexible dosing with aripiprazole could effectively reduce irritability associated with autistic disorder in Asian children and adolescents for up to 1 year. Our study also confirmed improvements in other secondary variables as in the antecedent short-term study in Korea (Kim et al. 2018).
The primary efficacy endpoint of this study was the mean change from baseline to the endpoint in Part 1 (week 12) in the caregiver-rated ABC-I subscale score and was discussed in our previous study (Kim et al. 2018). The ABC subscale scores, including social withdrawal, stereotypy, hyperactivity, and inappropriate speech, as measured by the ABC, as well as irritability, showed improvement during this Part 2 study up to 1 year. Our finding that the response to aripiprazole was maintained with continued treatment is consistent with previous long-term open-label study results in Japan and the United States (Marcus et al. 2011b; Ichikawa et al. 2018).
In a recent meta-analysis, aripiprazole was reported to be effective in the acute treatment of children and adolescents with autism spectrum disorder (ASD), including irritability, hyperactivity/noncompliance, inappropriate speech, and stereotypic behavior (Maneeton et al. 2018). This study complements the evidence that aripiprazole effectively reduced problematic behavior in Asian patients both acutely and in the long term.
The obsession and compulsion subscale scores of the CY-BOCS were also significantly improved by aripiprazole treatment. Some studies found improvement in the CY-BOCS within the first 8 weeks (Marcus et al. 2009) and the first 12 weeks (Kim et al. 2018). We found that the improvement in obsessive-compulsive symptoms was maintained over the 52-week treatment period, similar to the results of previous long-term studies (Marcus et al. 2011b; Ichikawa et al. 2018). Unlike other studies that used only the CY-BOCS compulsion scale, we also used the obsession scale, confirming a significant improvement in obsession.
Aripiprazole improved the quality of life measured by the Pediatric Quality of Life Inventory scale (Marcus et al. 2011b; Varni et al. 2012), and global functioning measured by the Clinical Global Impression (CGI) and Children's Global Assessment Scale (Ichikawa et al. 2018) in past studies. However, the evaluation of the improvement in core symptoms or adaptive function was somewhat limited. We additionally used the VABS to measure core symptoms and adaptive function improvement. Significant improvement in the communication and socialization subscales of the VABS appeared from 12 weeks and continued until 52 weeks. In a 10-year naturalistic longitudinal study on risperidone, social function measured by the Social Responsiveness Scale did not improve over time (Marrus et al. 2014). Also, in a systematic review of risperidone, there was no significant change in social or communicative function, although it effectively reduced stereotypy (McPheeters et al. 2011).
As can be seen from the results on the VABS and CGI, our study provides preliminary evidence that aripiprazole can improve core symptoms or adaptive functions in ASD (Kim et al. 2018) and that the improvement was maintained for up to 1 year. A recent study also used VABS for behavioral assessment, and it is expected that further results from that study will examine the effect of aripiprazole and risperidone on the core symptoms of ASD (DeVane et al. 2019).
The largest concentration of literature on parenting a child with ASD is focused on parental stress (Bonis 2016). In this study, scores on the parental distress, parent-child dysfunctional interaction, and difficult child subscales of the PSI-SF significantly improved during 52-week aripiprazole treatment, as well as 12 weeks of short-term treatment. The results of a long-term open-label study in the United States measured by the Caregiver Strain Questionnaire were consistent with our findings (Marcus et al. 2011b). Stress management for parents is critically important for improving the outcomes of children with developmental disabilities (Singer et al. 2007; Osborne et al. 2008). We observed that aripiprazole reduced parental stress, maybe as problem behaviors decrease, and we hope that it will be reference data for future studies that control parenting stress helps improve outcomes.
In terms of safety aspects, a total of 162 AEs were reported in 84.48% of the patients during the first 12 weeks of the study. Meanwhile, the total AEs reported in 52 weeks was 58.62%, which was lower than the rate at 12 weeks. The most commonly reported event at week 12 was weight increases in 25.86%, somnolence and sedation in 20.69% each, and nasopharyngitis in 13.79%. The most commonly reported event at week 52 was nasopharyngitis in 20.69%, and weight increases in 18.79%. The events reported in this study were comparable to other previously conducted aripiprazole studies (Marcus et al. 2009, 2011b; Owen et al. 2009; Ichikawa et al. 2017, 2018) and are listed on the current aripiprazole label.
Aripiprazole was better tolerated by patients over time. For example, the rate of somnolence in the Part 1 study was much higher than that in Part 2 (20.7% vs. 1.7%). Thus, aripiprazole was shown to be safe and well tolerated in the treatment of children and adolescents 6–17 years of age for up to 1 year. Also, as well as somnolence, the AE rate in our study (58.6%) was lower than the rate reported in a 52-week U.S. study (86.7%) (Marcus et al. 2011a). Racial differences may have influenced the results, but it is also necessary to consider different conditions such as the number of samples and the mean daily dosage of aripiprazole. The mean daily dose in this study (6.0 mg) was lower than that in the U.S. study (10.6 mg) (Marcus et al. 2011a).
In the results of two 52-week long-term aripiprazole studies recently reported in Japan, one used a mean daily dose of 7.2 mg and had an AE rate of 97.7% (Ichikawa et al. 2018). The other used an average dose of 2.2 mg and had an AE rate of 24.5% (Sugimoto et al. 2021). Although there were differences in the number of samples and study design, considering the results of the four studies described above, it is thought that the dosage had the most significant effect on AE incidence.
One SAE was a case of tonsillitis during the first 12 weeks of the study. In week 52, a total of three SAEs, including epiphysiolysis, seizure, and a suicide attempt, were reported in 1.72% (1/58 subjects, one case each). However, these SAEs were not ADRs. Among those, seizures and suicide attempts were rarely reported in earlier studies. One case of convulsion was reported in the U.S. study (Marcus et al. 2011a), and two cases of seizures were reported in a recent Japanese study (Sugimoto et al. 2021). Therefore, it is necessary to confirm whether these are observed in future studies.
Decreased serum prolactin levels with short-term aripiprazole treatment have been observed in previous studies (Marcus et al. 2009; Owen et al. 2009; Ichikawa et al. 2017; Kim et al. 2018). Longer-term aripiprazole treatment was also associated with an overall mean decrease in serum prolactin levels (Marcus et al. 2011a; Ichikawa et al. 2018). We obtained the same result, but its clinical consequences are unknown, as discussed in the previous study (Safer et al. 2013). Hyperprolactinemia is one of the common metabolic side effects of risperidone use (Almandil et al. 2013). Aripiprazole is usually used as monotherapy, but there is evidence that adjunctive aripiprazole treatment is effective and safe in treating antipsychotic-induced hyperprolactinemia (Meng et al. 2015). Therefore, we can consider the prolactin decrease as a solution to risperidone-induced hyperprolactinemia seen when risperidone is used for treating irritability in children and adolescents with autistic disorder.
Furthermore, there were no clinically significant changes in vital signs, EKGs, or EPSs at weeks 12 and 52. More importantly, there were no deaths or death-leading AEs, or suspected unexpected serious adverse reactions reported throughout the study. EPS-related AEs were not found in our study. This may be related to the use of low doses of aripiprazole (6.0 mg/day). In the previous U.S. 52-week open-label study, the mean daily dose of aripiprazole was 10.6 mg/day, and 48/330 (14.5%) subjects experienced EPS-related AEs (Marcus et al. 2011a). Of course, the difference in the number of samples must be taken into account.
This was the first study to demonstrate the long-term effectiveness and tolerability of aripiprazole in an Asian-focused multinational study. Our findings are noteworthy because we verified the long-term improvement and safety of aripiprazole at flexible doses for treating children and adolescents with irritability with autistic disorder in Asian populations.
However, there were several limitations to this study. First, compared to previous studies, the number of samples was relatively small. Second, although the open-labeled design can reflect the real-world clinical setting more, it can be a limitation because there is no comparison control group. Third, there could be a selection bias due to the 26 subjects in our study who previously received risperidone or aripiprazole.
Fourth, ADHD medication was allowed because the prescription was very important to maintain the patients' daily lives, and if removed would cause great distress. However, it was limited to cases where a stable dose was maintained for more than 4 weeks, and dose adjustment was not permitted during the study period, so the effect on the outcome variables is considered to be minimal. Fifth, except for the maintained ADHD medication, at some point during the study, 54% of subjects received concomitant medications, although it has been mainly used for a short period to control symptoms such as cough, cold, and rhinorrhea, which could affect the AE profile.
Despite these limitations, the strengths of this study are as follows: we were able to evaluate the improvement in core symptoms and adaptive function using the VABS. Our study, which continued for 52 weeks with an open label from the beginning, is a more genuine long-term study and may be closest to the actual clinical environment.
Conclusion
In conclusion, when aripiprazole was administered to Asian pediatric patients with autistic disorders and behavioral problems, general behavioral symptoms, including irritability and clinical severity, were significantly improved. Regarding safety, aripiprazole was tolerable, and the safety profile was favorable when aripiprazole was administered with tapering for 52 weeks. Therefore, up to 1 year, aripiprazole is effective and safe for Asian pediatric patients with autistic disorder.
Clinical Significance
This long-term open-label study demonstrated that flexible dosing with aripiprazole could effectively reduce irritability associated with autistic disorder in Asian children and adolescents both acutely and in the long term for up to 1 year. We additionally used the VABS to measure core symptoms and adaptive function improvement. Significant improvement in the communication and socialization subscales of the VABS appeared from 12 weeks and continued until 52 weeks.
Footnotes
Disclosures
This study was supported by Korea Otsuka International Asia Arab. No competing financial interests exist for authors.