
Abstract
Objective:
To describe the longer-term effectiveness, safety, and tolerability of open-label ziprasidone in children and adolescents with bipolar I disorder (BD-I).
Methods:
A subset of 23 participants aged 10–17 years, who were previously treated in a multi-site, 4-week randomized controlled trial received open-label ziprasidone (20–80 mg twice a day) for up to 26 weeks.
Results:
The most common adverse events (AEs) were fatigue (30%), somnolence (17%), and nausea (13%). Effects on weight, body mass index, and metabolic parameters (glucose, cholesterol, and triglycerides) were minimal. No participant had a Fridericia-corrected QT interval ≥ 460 msec or a change from baseline of ≥60 msec, and there were no cardiac-related AEs. Both the participants who continued ziprasidone and those who initiated ziprasidone in the open-label extension showed improvements in their symptoms of mania.
Conclusions:
The overall findings of the study are consistent with the accumulating knowledge on the safety profile of ziprasidone in the acute and long-term treatment of children and adolescents with BD-I, in the midst of a manic episode.
Introduction
Ziprasidone is an effective treatment for mania in adults with bipolar I disorder (BD-I) (Keck et al. 2003). A multi-site, double-blind (DB), randomized, 4-weeks, placebo-controlled study provided initial evidence to suggest that ziprasidone may have both acute efficacy and tolerability in youth aged 10–17 with BD-I suffering from mania (Findling et al. 2013). In part, based on the results of this first DB study, ziprasidone was approved by the European Medicines Agency for the treatment of this patient population (Zeldox SmPC 2019).
In a second randomized controlled trial (RCT) in youth with BD-I, ziprasidone again was shown to have both acute efficacy and acceptable tolerability in youth aged 10–17 years with mania (Findling et al. 2022). Since participants may receive treatment beyond several weeks, this report describes the longer-term safety, tolerability, and effectiveness of open-label ziprasidone in a subset of children and adolescents who had previously been treated in this second RCT.
Methods
Study design, population, and treatment
The DB, randomized 4-week parent study (protocol A1281198, NCT03768726) was conducted from May 2014 to May 2020 and was previously described in Findling et al. (2022). This extension study (protocol A1281201, NCT03768726) of the RCT was a single-arm, open-label, 26-week study designed to collect additional information on the safety, tolerability, and effectiveness of longer-term use of ziprasidone in the treatment of youth with BD-I. The open-label extension (OLE) was initiated after the acute clinical trial was ongoing for several years and was conducted from December 2018 to July 2020.
Due to the COVID-19 pandemic, enrollment into the acute study was halted in March 2020 and in the extension study, in June 2020. Both studies were approved by an Institutional Review Board/Ethics Committee before study enrollment and were conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki. All participants' parent(s)/guardian(s) provided written informed consent before any study-specific activity was performed.
Population
The details of the participants of the DB study have previously been published (Findling et al. 2022). For inclusion in the OLE study, participants were male and female youth aged 10–17 years, who were diagnosed with BD-I (current or most recent episode manic) based on The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) criteria, who completed the antecedent RCT (Findling et al. 2022) or otherwise qualified based on treatment response, were judged likely to benefit from continued antipsychotic therapy, and had no clinically significant safety concerns during their participation in the RCT (e.g., cardiac events or QT prolongation).
The exclusion criteria were similar to those in the DB study (Findling et al. 2022) and included any psychotic disorder other than BD-I, Substance Use Disorder, Autism Spectrum Disorder, Disruptive Mood Dysregulation Disorder, intellectual disability, and imminent risk of suicide or homicide.
Treatment
Participants who continued into this OLE study entered an initial 2-week DB transition period designed to protect the blind of the RCT. Participants who were on active ziprasidone in the DB study continued active ziprasidone in this study, and those originally on placebo were switched to active ziprasidone. Participants weighing <45 kg who initiated ziprasidone in the OLE were titrated over the 1st week of the study in 20 mg increments to a final total daily dose of 60 mg (given as a divided dose in the AM and PM), and participants weighing ≥45 kg were titrated over the first 2 weeks of the study to a final total daily dose of 120 mg (also given as a divided dose). At the end of the transition period, all participants received flexibly dosed open-label ziprasidone (Fig. 1).
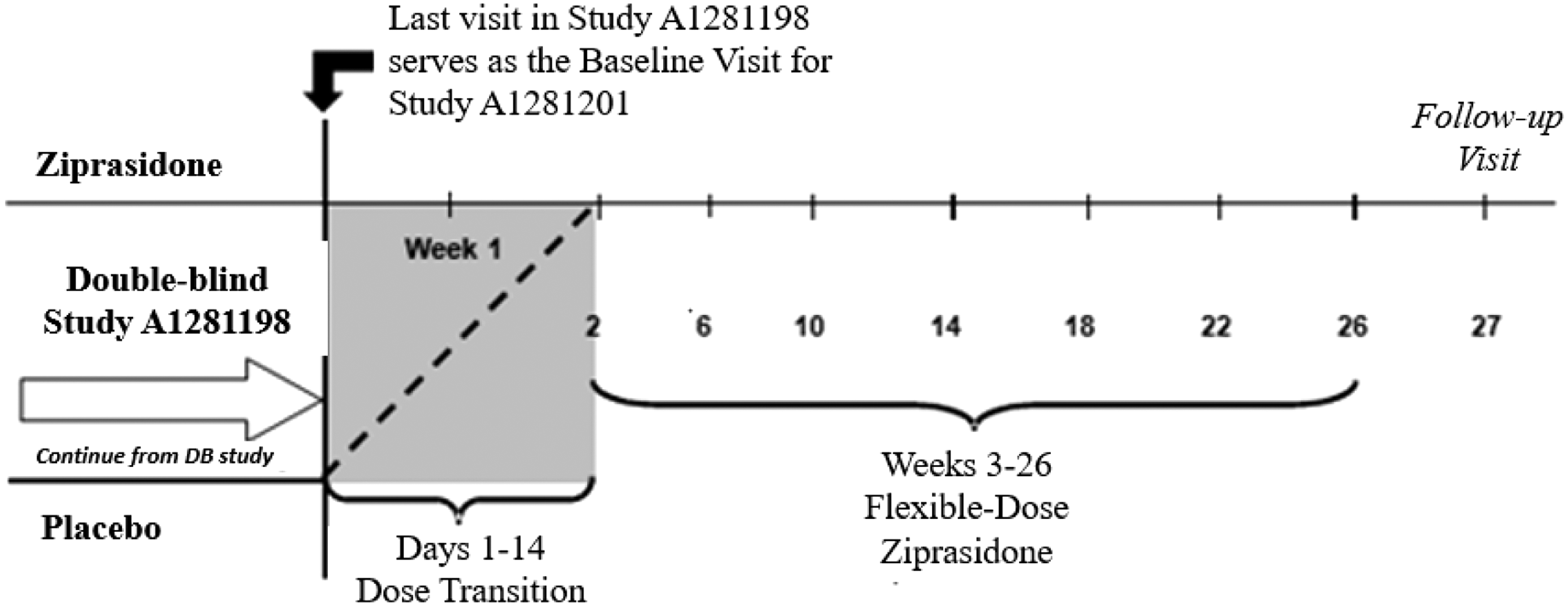
Study schema.
Central nervous system active agents (including attention-deficit/hyperactivity disorder medications such as stimulants, alpha-2 adrenergic agonists, and atomoxetine) were prohibited during the study. Lorazepam or a comparable benzodiazepine was permitted (2 mg/day) for anxiety or agitation. In addition, lorazepam, diphenhydramine, melatonin, or zolpidem could be used for insomnia; benztropine, benzhexol, and other anticholinergics or propranolol could be used to treat extrapyramidal symptoms if needed.
Assessments
Safety was the primary objective of this OLE study and was assessed by adverse events (AEs), clinical laboratory testing, physical examinations, electrocardiogram (ECG), and monitoring of vital signs, weight, body mass index (BMI), and BMI-z scores (which are standardized for age and sex of the participant). The Columbia Suicide Severity Rating Scale (C-SSRS; Posner et al. 2008), The Child Depression Rating Scale Revised (CDRS-R; Poznanski et al. 1985), and movement disorder scales (Simpson-Angus Rating Scale [SARS]; Simpson and Angus 1970, Barnes Akathisia Rating Scale [BAS]; Barnes 1989, Abnormal Involuntary Movement Scale [AIMS]; Guy 1976) were administered at every visit.
The CDRS-R was included as a safety assessment (instead of an efficacy measure) to monitor participants for the risk of being switched from a manic state into a depressed state. The secondary efficacy outcomes were changed from baseline to week 26 in Young Mania Rating Scale (YMRS; Fristad et al. 1995; DelBello et al. 2002), Clinical Global Impression of Severity (CGI-S; Guy 1976) score, and Clinician-Rated Global Assessment (CGAS; Shaffer et al. 1983).
Statistical analysis
The number of participants entering this OLE study was determined by the number of participants electing to continue treatment after completing or withdrawing from the preceding DB study. As the OLE was an open-label noncomparative study, no inferential statistical analyses were performed. Quantitative variables were summarized by descriptive statistics (mean, standard deviation [SD], 95% confidence interval), and qualitative variables were summarized in frequency tables. Summaries included data for all participants who took at least one dose of investigational product in this OLE study. For efficacy endpoints, baseline visit for the OLE study was considered the final visit of the DB study (week 4 or early termination visit from RCT).
For safety assessments, for those participants who were randomized to placebo in the DB study, baseline values were the last observation before initiating dosing in the OLE study, and for those participants who were randomized to ziprasidone in the DB study, baseline values were the values from the baseline visit in the RCT.
Results
Participant disposition
Of the 171 participants who entered the RCT, 23 entered the OLE; 12 (52.2%) of the 23 participants completed the 26-week study, and 11 participants (48%) discontinued. The mean duration of ziprasidone treatment during the OLE was 124 days. In terms of retention, 4 participants received <28 days of ziprasidone; 3 received >29 to 60 days; 6 received from 61 to 180 days; and 10 received ≥180 days of treatment.
Reasons for discontinuation were AEs (4 participants, 17.4%) and withdrawal of consent (6 participants, 26.1%); one participant was lost to follow-up. The AE's leading to study discontinuation each occurred in only 1 of the participants who discontinued and included: fatigue, memory impairment, sedation, and somnolence.
At baseline of the OLE, the mean (SD) age of the participants was 14.1 (2.07) years. There were 9 participants in the 10–13 year age bracket and 14 in the 14–17 year age bracket. The majority were white 18 (78.3%). Twelve were female (52%). Mean daily modal dose of ziprasidone during weeks 2–26 for participants <45 kg (N = 6) was 66.67 mg and for participants ≥45 kg (N = 17), it was 91.76 mg. In the <45 kg treatment group, 3 participants received total daily doses <80 mg/day and 3 participants received total daily doses ≥80 mg/day. In the ≥45 kg treatment group, 5 participants received total daily doses <80 mg/day; 1 received a total daily dose ranging from 80 to <100 mg/day; and 11 received total daily doses ranging from 100 to <160 mg/day.
Safety
All-causality treatment-emergent adverse events (TEAEs) were reported in 19 participants (82.6%). Most TEAEs were mild or moderate in intensity, with two severe events reported (suicidal ideation and fatigue). Nervous system disorders were the most common class of TEAEs and occurred in 43.5% of participants. Consistent with the RCT, the most commonly reported TEAEs were fatigue (30%), somnolence (17%), and nausea (13%) (Table 1; Supplementary Table S1). One participant reported two SAEs of suicidal ideation (non-fatal) and aggression that were not considered to be related to ziprasidone by the investigator.
Treatment-Emergent Adverse Events (≥5%; Safety Population)
Subjects were counted only once per event.
Akathisia, muscle contractions involuntary, muscle rigidity, muscle spasms, muscle twitching, musculoskeletal stiffness, oromandibular dystonia, psychomotor hyperactivity, restlessness, tremor, and trismus.
AEs, adverse events.
All the TEAEs of fatigue, nausea, and extrapyramidal symptoms (dystonia, akathisia, and musculoskeletal stiffness) occurred in participants who initiated ziprasidone for the first time in the extension study (placebo/ziprasidone, N = 13). Transitory AEs occurred during the crossover for those participants who were in the placebo arm during the titration to the weight-based target dose; the AEs largely resolved with continued treatment at the target dose or a temporary reduction in dose.
The CDRS-R scores tended to decrease from baseline over the study for all participants. Three participants had CDRS-R scores ≥40 at some point during study participation (Poznanski and Mokros 1996). None experienced a sustained depressive mood.
No participants had a Fridericia-corrected QT (QTcF) interval ≥460 msec or a change from baseline of ≥60 msec during the OLE. Mean QTcF change from baseline (defined as the QTcF on day 1 of the parent placebo-controlled study) at the end of treatment (week 26) was 6.7 msec (range −14.0 to 49.0). No cardiac-related TEAEs or AEs indicative of QT prolongation were reported.
Elevated prolactin (>1.1 × Upper Limit of Normal and >1.5 × baseline value) was observed in 8 of the participants (5 females and 3 males). The elevations were not viewed as being clinically significant and were not reported as or associated with AEs. The average final prolactin value was 42.2 mcg/L (range 11.3 to 68.7 mcg/L) in the 5 females and 17.6 mcg/L (range 13.1 to 22.6 mcg/L) in the 3 males.
The mean (SD) baseline weight at the start of the OLE was 60.38 kg ±18.37, and the mean weight gain from baseline to week 26 (end of treatment) was 2.08 kg ±3.81. The mean baseline BMI and BMI-z scores were 2.91 kg/m2 ± 4.79 and 0.85 ± 1.02. Mean changes in BMI and BMI-z scores from baseline to the end of treatment were 0.57 kg/m2 ± 1.56 and 0.14 ± 0.28. One participant had a BMI z-score change of ≥1 from the OLE baseline to week 26 (end of treatment).
Baseline and changes from baseline to week 26 (end of treatment) in metabolic parameters (obtained in 21 participants) were: fasting glucose (mg/dL), 89.0 ± 11.14 and 3.4 ± 9.57; total fasting cholesterol (mg/dL), 156 ± 24 and 5 ± 21; and fasting triglycerides (mg/dL), 93 ± 53 and 7 ± 40. There were no AEs of elevated glucose or insulin assessed as related to ziprasidone by the investigator.
Consistent with the RCT, mean total scores of the movement disorder scales (SARS, BAS, AIMS) were low and generally similar at all visits. The AIMS Global Judgment results were normal for all participants except for a single participant who reported minimal abnormal movements at week 2 of the study.
Secondary effectiveness
Descriptive statistics for the YMRS total score showed that participants who continued ziprasidone treatment into the extension study (ziprasidone/ziprasidone, N = 10) maintained or improved their response. The mean (SD) baseline and mean (SD) change from baseline at the end of treatment (week 26) was 12.7 (8.31) and −1.9 (4.85). Participants who initiated ziprasidone in the extension study (placebo/ziprasidone, N = 13) showed improvement in their symptoms of mania based on the YMRS ratings. The mean (SD) baseline and mean (SD) change from baseline at end of treatment in the placebo/ziprasidone group was 12.0 (5.46) and −6.4 (5.50).
The mean (SD) baseline and mean (SD) change from baseline to week 26 for CGI-S scores was 2.7 (1.25) and −0.1 (1.25) in the ziprasidone/ziprasidone group and 2.5 (0.97) and −0.5 (1.05) in the placebo/ziprasidone group, indicating an improved response. The mean (SD) baseline and mean (SD) change from baseline to week 26 in CGAS scores was 68.2 (16.4) and 1.6 (11.6) in the ziprasidone/ziprasidone group and 67.7 (13.28) and 7.2 (10.13) in the placebo/ziprasidone group, respectively.
Discussion
This OLE study provides additional evidence of the safety, tolerability, and efficacy of ziprasidone in the long-term treatment of youth with BD-I and adds to the previously published long-term safety study (Findling et al. 2013a).
The safety and tolerability data from the current study are consistent with the long-term safety and tolerability profile previously reported by Findling et al. (2013a). No new safety signals were identified, and there were no trends associated with the longer treatment duration.
Special safety assessments (e.g., movement disorder scales, C-SSRS, and CDRS-R) generally showed no meaningful changes in scores over time. No evidence of increased suicidal ideation/behavior or depression was found with longer-term administration of ziprasidone. There were no new or unexpected safety findings in laboratory results. Ziprasidone was not associated with any meaningful changes in weight, BMI, BMI z-score, or metabolic parameters. No cardiac AEs or AEs related to ECG abnormalities were reported. No participants had QTcF intervals ≥460 msec or a change from baseline of ≥60 msec during the study.
The major limitations of this study include the relative brevity of the treatment duration and the small number of participants that does not allow for the detection of uncommon side effects. The principal reasons that so few subjects entered the OLE were: (1) the fact that the extension study was not initiated until the placebo-controlled study had been ongoing for several years and (2) the interruption of study recruitment in March 2020 due to the onset of the COVID pandemic.
An extensive review of the literature failed to reveal any head-to-head studies (short or longer term) directly testing the safety and efficacy of ziprasidone compared with other medications for the treatment of mania in children and adolescents 10–17 years old. Longer-term (>3 months) studies of the safety and tolerability of second-generation antipsychotics (SGAs) in pediatric bipolar disorder (BPD) are limited in number. However, open-label, monotherapy, longer-term studies are available for olanzapine (Detke et al. 2016), quetiapine (Findling et al. 2013b), and asenapine (Findling et al. 2016), as well as ziprasidone (Findling et al. 2013a) (Supplementary Table S2).
The studies of olanzapine (Detke et al. 2016) and quetiapine (Findling et al. 2013b) enrolled children and adolescents with either schizophrenia or BPD. In addition, a dose-ranging, placebo controlled, 30-week study provides longer term data on the safety and efficacy of aripiprazole in the treatment of children and adolescents with mania (Findling et al. 2012).
Collectively, these studies range in duration from 26 to 52 weeks and provide systematic assessments of weight gain, change in BMI, and cardiometabolic and endocrine effects of the SGAs studied, although differences across studies in the choice of metrics and how the data were analyzed and presented present some challenges in drawing comparisons among them. No longer-term study of risperidone monotherapy in the treatment of pediatric BD-I or schizophrenia was identified by our team. However, in shorter-term studies of these disorders and longer-term studies in autism, risperidone has been associated with moderate to severe weight gain and elevated prolactin (De Hert et al. 2011; Fraguas et al. 2008; Peuskens et al. 2014).
Inspection of the results of the available longer-term studies (Supplementary Table S2) reveals that in general the effects of longer-term treatment with these drugs shows a similar heterogeneity of effects among the different drugs as was observed in the shorter treatment studies. In these studies, ziprasidone again appears to be associated with the lowest mean overall weight gain from baseline (2.08 ± 3.81 kg); quetiapine (3.3 ± 9.1 kg in schizophrenia patients and 4.0 ± 5.2 kg in bipolar patients); asenapine (3.5 ± 5.8 kg) and aripiprazole (6.5–6.6 kg) with intermediate weight gain, and olanzapine with the greatest weight increase (9.6–12.1 kg).
Olanzapine was also associated with a marked increase from baseline in mean BMI (2.8–3.6 kg/m2). In studies reporting this metric, the proportion of patients with a ≥7% increase from their baseline weight was commensurate with the increases in overall mean weight: quetiapine: 29.1% of schizophrenia patients and 41.3% of bipolar patients; asenapine: 34.8%; olanzapine: 40%. Among the children and adolescents treated with aripiprazole for up to 30 weeks, 8.8%–13.6% shifted to an obese BMI percentile during the course of the study (Findling et al. 2012).
Mean changes from baseline in metabolic parameters were unremarkable for ziprasidone, aripiprazole, and asenapine (Supplementary Table S2). However, olanzapine was associated with a marked increase in triglycerides (26.55 ± 8.85 to 35.4 ± 8.85) (Detke et al. 2016), and 8.4%–11.9% of children and adolescents treated with quetiapine had triglyceride levels ≥200 mg/dL at the end of treatment whereas 13.4%–16.5% had high-density lipoprotein, HDL cholesterol levels ≤40 mg/dL (Findling et al. 2013).
Although changes in overall laboratory metabolic measures were minimal in the children and adolescents treated with asenapine for up 50 weeks, a total of 18 patients met criteria for new onset Metabolic Syndrome during the course of the study, leading the study authors to recommend metabolic monitoring in pediatric patients initiating treatment with asenapine (Findling et al. 2016).
In these longer-term studies, aripiprazole and olanzapine were associated with mean decreases from baseline in serum prolactin, whereas quetiapine and asenapine either had no effect or caused a mild decrease. In the largest ziprasidone longer-term study (N = 162 participants treated for up to 26 weeks; Findling et al. 2013a), there was a modest increase in serum prolactin (1.9 ± 8.5 ng/mL) and 7% of patients had an elevated serum prolactin (>1.1 × upper limit of normal) at some point in the study.
Change in mean QTcF was reported only for quetiapine (−0.3 ± 25.2 msec for schizophrenia patients, −4.1 ± 29.4 msec for BPD patients), olanzapine (0.9 to 2.0 ± 1.6 msec) and ziprasidone (5.3 ± 16.0 msec and 6.7 msec, range: −14.0 to 49 msec).
Ziprasidone does not have regulatory approval for use in children and adolescents with bipolar I mania in the United States, but it is approved for acute treatment in the European region. However, considering the limited therapeutic options for children with BD-I, it is beneficial to offer clinicians information regarding ziprasidone studies in long-term treatment of the disorder. Despite access to treatment, up to 40%–60% of bipolar youth continue to have an inadequate response or fail to achieve remission given the complexity of the disorder (Smarty and Findling 2007; Nandagopal et al. 2009; Stepanova and Findling 2017).
Several cardiac risk factors that would preclude ziprasidone treatment include a pre-existing cardiac arrhythmia or a long QTc interval (either congenital or acquired) (Zeldox SmPC 2019).
Conclusion
Data from this 26-week open-label, single-arm extension that enrolled a small number of participants from the randomized, DB, placebo-controlled, phase 3 study reported recently by Findling et al. (2022) add to the accumulating knowledge (Findling et al. 2013a, 2018; Stepanova and Findling 2017; Sun et al. 2019) on the safety, tolerability, and efficacy of ziprasidone in the acute and long-term treatment of children and adolescents aged 10–17 years with BD-I in the midst of a manic episode.
Footnotes
Acknowledgments
The authors would like to acknowledge Rekha Raghuram MSc. from Viatris, Bengaluru, India for her medical writing assistance in developing this manuscript.
Authors' Contributions
All the authors confirm that they were part of this study and have played a significant role from conceptualization of the study to the manuscript preparation. Authorship for this study was considered in accordance with the ICMJE and COPE guidelines of authorship. All the authors approved the final version of the manuscript along with the author order as presented in the manuscript.
P.C. and R.L.F. contributed to the study conceptualization, design, recruitment of participants, data collection, data analysis, result interpretation, and manuscript writing. S.A., M.B., and C.I. contributed to design, data collection, data analysis, and result interpretation. P.A. contributed to the analytical plan and development of the study protocol. Y.R. took part in data analysis, results interpretations, and manuscript writing.
Disclosures
R.L.F. receives or has received research support, acted as a consultant, and/or has received honoraria from Acadia, Adamas, Aevi, Afecta, Akili, Alcobra, Alkermes, Allergan, American Academy of Child and Adolescent Psychiatry, American Academy of Neurology, American Psychiatric Press, Arbor, Axsome, Gedeon Richter, Idorsia, Intra-Cellular Therapies, Luminopia, Lundbeck, MedAvante-ProPhase, MJH Life Sciences, NIH, Neurim, Otsuka, PaxMedica, PCORI, Pfizer, Physicians Postgraduate Press, Q BioMed, Receptor Life Sciences, Roche, Sage, Signant Health, Sunovion, Supernus Pharmaceuticals, Syneos, Syneurx, Takeda, Teva, Tris, Validus, and Viatris. P.C., M.B., P.A., and C.I. are all full-time employees of Pfizer, Inc. Y.R. is a full-time employee of GE Healthcare.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.