
Abstract
Background:
Oxcarbazepine is thought to be better-tolerated and less susceptible to drug–drug interactions than its predecessor, carbamazepine. Genetic testing for HLA-B*15:02 is recommended in specific populations to identify those at high risk of severe hypersensitivity reactions; however, other pharmacologic and pharmacogenetic factors that can impact drug disposition may be involved.
Methods:
We present a case of an 8-year-old boy treated with oxcarbazepine who developed drug reaction with eosinophilia and systemic symptoms (DRESS) with Stevens–Johnsons syndrome overlap and was negative for HLA-B*15:02. We review the extant literature related to oxcarbazepine disposition, and potential pharmacogenetic variants in aldoketoreductase 1C (AKR1C)2–4 that may contribute to this risk.
Results:
Genetic variability in oxcarbazepine disposition pathways may contribute to tolerability and toxicity, including the development of hypersensitivity reactions.
Conclusions:
While preemptive genetic testing for HLA-B*15:02 in individuals of Asian ancestry is recommended to prevent severe hypersensitivity reactions to oxcarbazepine, oxcarbazepine concentrations and AKR1C variation may contribute to the risk of severe adverse reactions. We provide recommendations for future study to elucidate whether these individual factors are important for reducing the risk of severe adverse events.
Introduction
Oxcarbazepine, a keto derivative of carbamazepine, is approved by the U.S. Food and Drug Administration (FDA) for pediatric epilepsy but is used off-label in children and adolescents to treat affective symptoms and agitation (Douglas et al., 2013; Ionova et al., 2022; Morrow et al., 2020; Novartis, 2017; Sourbron et al., 2023; Wagner et al., 2006). It is considered a safer alternative to carbamazepine due to fewer drug–drug interactions and an improved side effect profile. However, serious, potentially life-threatening hypersensitivity can occur, specifically Stevens–Johnson syndrome/Toxic Epidermal Necrosis (SJS/TEN) and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS). SJS/TEN involves immune-mediated necrosis to skin and mucous membranes resulting in skin detachment over large areas of the body and risk of infection, hypovolemic shock, and multiple organ dysfunction. DRESS is associated with multiple organ dysfunction, prolonged hospital stays, and increased health care costs (Wolfson et al., 2019; Yang et al., 2019a).
Individuals with a genetic marker, HLA-B*15:02, are at increased risk of SJS/TEN with carbamazepine, and the risk extends to structurally similar aromatic anticonvulsants, such oxcarbazepine (Phillips et al., 2018). Indeed, the Clinical Pharmacogenetic Implementation Consortium (CPIC) recommends against its use in naive individuals with an HLA-B*15:02 allele due to increased risk of SJS/TEN (Phillips et al., 2018) and the FDA recommends consideration of preemptive genetic testing before oxcarbazepine initiation in individuals with ancestry known to have a higher frequency of the HLA-B*15:02 genotype like Han Chinese (2%–12%), Thai (8%), Philippines/Malaysian (15%), Korean (2%), and Indian (6%) (Novartis, 2017).
HLA-B*15:02 is relatively rare (<1%) in individuals of non-Asian descent (e.g., African, European, Indigenous American, Hispanic, and Japanese) (Zhou et al., 2021). Further, the evidence for oxcarbazepine-induced SJS/TEN and HLA-B*15:02 is weaker than for carbamazepine (Chen et al., 2017; Hung et al., 2010). Although still a notable safety signal, oxcarbazepine is associated with a lower risk of SJS/TEN relative to carbamazepine (Xu et al., 2021). The association between HLA-B*15:02 and other oxcarbazepine-induced hypersensitivity, specifically DRESS, is not well known, particularly in children and adolescents (Jantararoungtong et al., 2021).
Given these mixed data, what constitutes “high risk” for oxcarbazepine-treated patients is unclear. Herein, we describe a pediatric patient who was HLA-B*15:02 negative and who developed DRESS with SJS overlap and discuss additional pharmacogenetic and pharmacologic factors that may contribute to oxcarbazepine-associated severe hypersensitivity and toxicity.
Case Report
A, an 8-year-old boy with attention-deficit/hyperactivity disorder, an unspecified mood disorder, and seasonal allergies was treated with extended-release d-methylphenidate (0.8 mg/kg = 20 mg each morning). Self-reported race and ethnicity was white, non-Hispanic. Two months after beginning extended-release d-methylphenidate, oxcarbazepine was added for mood stabilization. Eleven days after beginning oxcarbazepine, he developed pruritic, erythematous macules on the dorsal and plantar surfaces of his feet, followed by fever (39°C), malaise and the development of erythematous macules, and edematous papules on his arms, legs, and trunk the following day.
He was taken to the emergency department on his physical examination; in the emergency department, he had bilateral conjunctival injection and erythema of the lip with small palatal erosions. He did not report any myalgia, arthralgia, sore throat, photophobia, cough, congestion, vomiting, diarrhea, or dysuria. Etanercept 25 mg SQ and intravenous methylprednisolone 2 mg/kg were administered in conjunction with IV diphenhydramine 25 mg, PO acetaminophen 10 mg/kg, and a normal saline bolus of 20 mL/kg. His complete blood count, hepatic profile, prothrombin time, international normalized ratio were within normal limits; eosinophils were mildly elevated at 0.58, neutrophils were elevated (9.57), and his laboratory studies were further remarkable for a sedimentation rate (14, peaked at 17), C reactive protein (1.3, peaked at 2.3), D-dimer (0.61, normal ≤0.49), and lymphocytes (0.91, normal 1.5–4.9) (see Supplementary Material for Full Lab Results). He was admitted and d-methylphenidate and oxcarbazepine were held. IV methylprednisolone was continued at 2 mg/(kg·d) with a gradual taper to oral prednisone. He received hydroxyzine 13 mg QID as needed for pruritis and IV pantoprazole 25 mg/day for 2 days, followed by oral omeprazole (20 mg daily). Over 5 days, his rash improved without further involvement of mucus membranes. He did have a mild elevation of AST/ALT that continued to trend up upon discharge. Extended-release d-methylphenidate was resumed upon discharge.
He was negative for HLA-A*31:01, HLA-B*15:02, HLA-B*57:01, and HLA-B*58:01. He was diagnosed with DRESS with mild SJS overlap due to fever, eosinophilia, and rash with mucous membrane involvement. As an outpatient, oral methylprednisolone was tapered from 40 mg daily to 5 mg every other day for 2 months. Serial complete blood counts with differentials and hepatic profiles normalized over 2 months. Finally, the parent and patient provided consent and assent to publish this report.
Oxcarbazepine Disposition
Oxcarbazepine has an oral bioavailability of 99% and following absorption, undergoes reduction in the liver to its primary metabolite licarbazepine (MHD for monohydroxy derivative), by the cytosolic enzyme family, aldoketoreductase 1C (AKR1C) (Fig. 1) (Ichida et al., 2023; Malátková et al., 2014). Both oxcarbazepine and MHD contribute to the activity of the drug by inhibiting voltage-sensitive sodium channels, stabilizing hyperexcited neuronal membranes, inhibiting repetitive firing, and decreasing the propagation of synaptic impulses (Lexicomp, Waltham, MA). Relative to oxcarbazepine, MHD peaks later, reaches plasma concentrations >10-fold higher, and is eliminated more slowly.
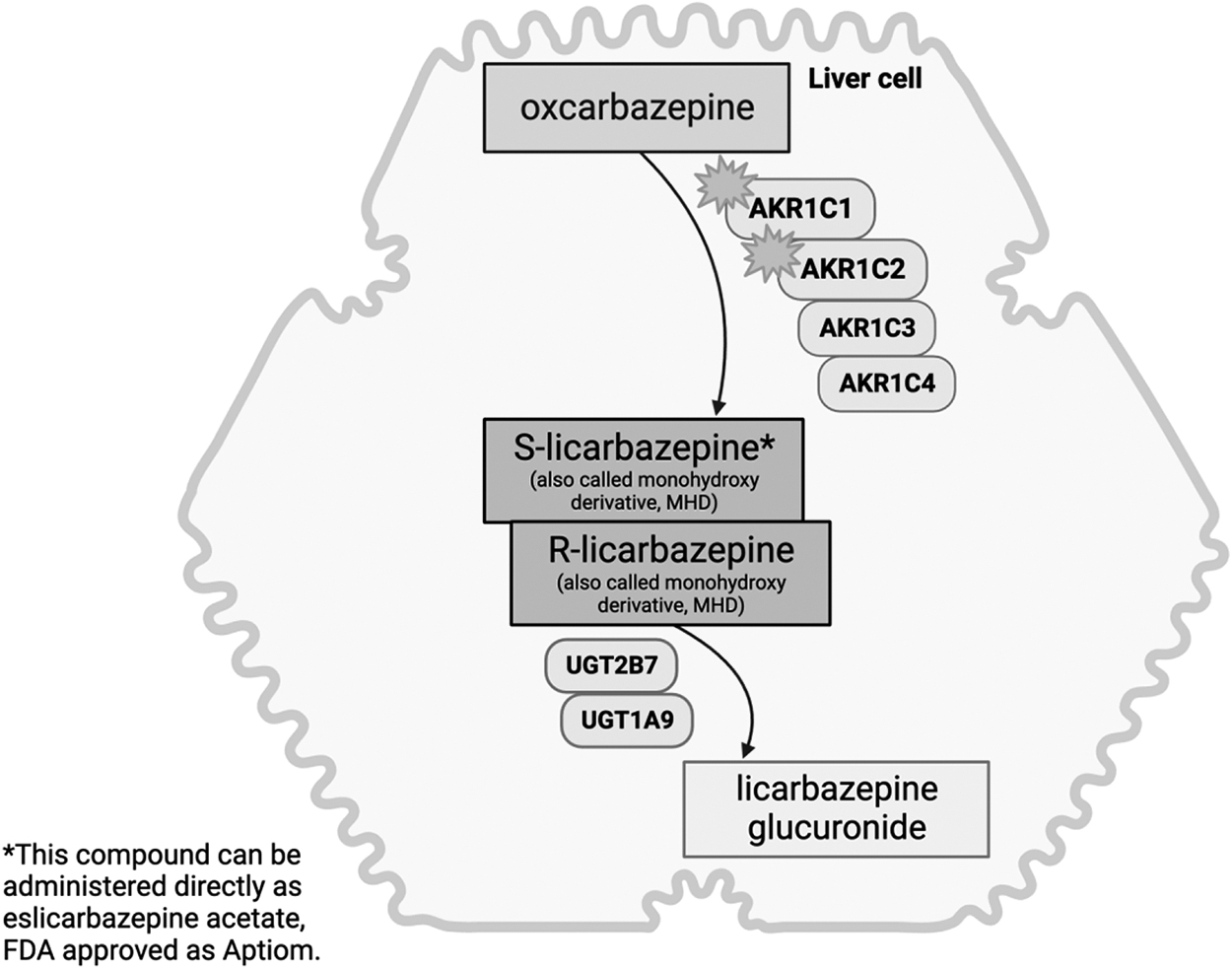
Drug metabolizing enzymes involved in oxcarbazepine disposition in the liver.
AKR1C1 and AKR1C2 are likely the primary enzymes responsible for the formation of MHD at clinically relevant oxcarbazepine exposures (plasma Cmax 4–24 μM) based on in vitro enzyme affinity (Km) (González-Esquivel et al., 2000; Malátková et al., 2014). However, hepatic oxcarbazepine concentrations are unknown and presumed to be higher than plasma, suggesting the possible contribution of AKR1C3 and AKR1C4 as well (Malátková et al., 2014). While AKR1C4 is exclusively expressed in the liver, AKR1C1–3 are expressed in the brain and adipocytes (Uhlén et al., 2015). Interestingly, AKR1C1–3 enzymes in adipocytes have demonstrated the ability to metabolize medication (Sheng et al., 2017).
MHD is primarily metabolized by glucuronidation through UDP-glucuronosyltransferase (UGT) enzymes. There is also some back-transformation from MHD to oxcarbazepine. Although not metabolized by CYP450, oxcarbazepine is a weak inhibitor of CYP2C19 and a weak inducer of CYP3A4. Further, the efflux transporter, P-glycoprotein (P-gp) is involved in oxcarbazepine exposure by reducing entry and enhancing removal of oxcarbazepine and MHD from the small intestine and brain (Fortuna et al., 2012; Marchi et al., 2005). The s-enantiomer of the MHD (s-licarbazepine) is not a substrate for P-gp, a favorable quality for drugs targeting the brain where P-gp decreases blood–brain barrier entry (Fortuna et al., 2012). Infants and children have higher weight-normalized clearance and volume of distribution of MHD than adults suggesting the need for increased doses for young children. Volume of distribution is large for both oxcarbazepine and MHD, suggesting vast distribution in extravascular tissues (Chen et al., 2021; Rodrigues et al., 2017).
Oxcarbazepine Concentrations and Toxicity/Tolerability
Most pediatric studies of oxcarbazepine focus on the active metabolite, MHD (Chen et al., 2021; Rey et al., 2004) and for therapeutic drug monitoring, MHD is targeted with a recommended range of 3–35 mg/L to avoid toxicity (Bring and Ensom, 2008; Patsalos et al., 2008). Understanding the link between oxcarbazepine systemic exposure and tolerability or toxicity is limited by the lack of available data. Several studies have evaluated oxcarbazepine dose and MHD exposure and treatment-emergent adverse reactions. However, to date, the association between oxcarbazepine dose and MHD exposure or MHD exposure and adverse events is unclear.
Common adverse reactions to oxcarbazepine, such as dizziness, nausea, vomiting, nystagmus, ataxia, and diplopia, typically manifest within 4 hours of dosing and usually subside within 24 hours (Kim et al., 2013; Kim et al., 2012). Plasma oxcarbazepine concentrations peak near 2 hours and quickly fall by 24 hours. In contrast, plasma MHD concentrations steadily rise throughout the day, reaching levels 38-fold higher than oxcarbazepine by 24 hours postdose (Kim et al., 2012; Novartis, 1999a, b). The onset and duration of the most common side effects appear more aligned with oxcarbazepine plasma concentrations, and data do not suggest a relationship between MHD levels and treatment-emergent side effects. Further, there is evidence of successful desensitization to oxcarbazepine following oxcarbazepine-induced rash in HLA-negative individuals, accomplished by low initial exposure with slow titration over time (Lee et al., 2017; Lee et al., 2014). Collectively, these findings suggest that oxcarbazepine levels, rather than MHD, may be responsible for treatment-emergent side effects. Unfortunately, most studies report MHD concentration rather than oxcarbazepine levels making it difficult to investigate with the data available.
The FDA approval of the s-enantiomer of MHD, formulated as eslicarbazepine acetate, provides an opportunity to observe differences in treatment-emergent side effects between oxcarbazepine and MHD (Sunovion, 2013). Oral eslicarbazepine leads to fourfold lower oxcarbazepine exposure and is associated with reduced treatment-emergent side effects and less discontinuation compared with oxcarbazepine (Zhang et al., 2022). Patients who discontinued oxcarbazepine due to side effects have generally tolerated eslicarbazepine, with only 1 in 15 of oxcarbazepine-intolerant patients discontinuing eslicarbazepine due to side effects (Mäkinen et al., 2017; Rocamora et al., 2020). Further, eslicarbazepine has a lower incidence of severe hypersensitivity reactions compared to oxcarbazepine, although the association with HLA genotype has not been fully explored (Rogin et al., 2020).
Other Pharmacogenetic Variants
Beyond the immune-mediated hypersensitivity associated with HLA-B*15:02, genetic variation in oxcarbazepine disposition (e.g., drug metabolizing enzymes and drug transporters) may impact tolerability and toxicity (Fig. 1).
Genes involved in the regulation (HNF4α), drug transport (ABCB1, ABCC2), phase 2 drug metabolism (UGT1A9, UGT2B7), and drug action (SCN1A, SCN2A) of oxcarbazepine have been investigated (Ma et al., 2015; Shen et al., 2017; Yang et al., 2019b). Some studies show no association between ABCB1 variation and systemic MHD exposure (Lin et al., 2019), while others demonstrate a relationship between ABCB1, ABCC2, and MHD concentration/dose ratio or maintenance dose (Ma et al., 2015; Shen et al., 2017; Yao et al., 2022). Other studies have found no clear association between MHD systemic exposure or plasma concentration and genetic variation in HNFα, UGT2B7, UGT1A9, or ABCB2 (Lin et al., 2019; Shen et al., 2017; Zhao and Meng, 2022).
ABCB1 rs1045642 and UGT2B7 rs7432366 were associated with oxcarbazepine efficacy in epilepsy, while HNFα rs2071197, ABCC2 rs2273697, and SCN2A rs17183814 were not (Ma et al., 2015; Shen et al., 2017; Yang et al., 2019b). The role of genetic variation in other drug metabolizing enzymes on oxcarbazepine exposure was recently reviewed (Zhao and Meng, 2022). However, there are no data evaluating the impact of AKR1C genetic variation on oxcarbazepine exposure, tolerability, or toxicity. There are several genetic variants in AKR1C family that are associated with reduced enzyme activity that could contribute to altered oxcarbazepine concentrations (Table 1); however, these have yet to be explored.
AKR1C Genetic Variants with Observed Functional Consequences
AKR1C, aldoketoreductase 1C; MAF, minor allele frequency; SNP, single nucleotide polymorphism.
Conclusion
Oxcarbazepine is associated with severe, potentially life-threatening hypersensitivity and targeted preemptive genetic testing for individuals of certain ancestry is recommended to prevent these reactions. However, patients may endorse multiple race/ethnicities and self-report and genetic ancestry may be discrepant (Banda et al., 2015; Mersha and Abebe, 2015), making decisions regarding ancestry-based preemptive genetic testing nebulous. If a universal preemptive HLA-B*15:02 approach is employed, considerations include the potential delay in obtaining results and subsequent delays in treatment, testing cost, and expectations of actionability. In this case, if the primary objective was to reduce risk of severe hypersensitivity reactions, preemptive testing would not have discouraged oxcarbazepine use.
This report raises the possibility that evaluating oxcarbazepine concentrations may provide insight into potential exposure-related toxicity otherwise missed if only MHD concentrations are obtained. The availability of oxcarbazepine concentrations early in treatment would provide an opportunity to understand the association between oxcarbazepine exposure and risk of severe hypersensitivity reactions or treatment-limiting adverse reactions. While a target range currently exists for MHD, characterization of oxcarbazepine concentrations potentially lays the foundation for therapeutic drug monitoring of both parent (oxcarbazepine) and metabolite (MHD).
Further, as access to pharmacogenetic testing increases, we have the opportunity to probe genetic variants relevant to oxcarbazepine metabolism (e.g., AKR1C family) in addition to those involved in immunogenicity (e.g., HLA-B). Pairing oxcarbazepine exposure with a broader examination of pharmacogenetic variation may elucidate patient-specific factors that contribute to the risk of hypersensitivity and other adverse reactions and, ultimately, lead to safer use.
Clinical Significance
Oxcarbazepine is associated with severe, potentially life-threatening hypersensitivity reactions, not solely explained by the presence of HLA-B*15:02. Measuring oxcarbazepine concentrations, in addition to metabolite (monohydrate derivative) concentrations, paired with a broader pharmacogenetic inquiry may elucidate patient-specific factors that contribute to altered oxcarbazepine exposure and risk of hypersensitivity and other adverse reactions.
Footnotes
Authors' Contributions
S.L.S.: conceptualization (lead), investigation, resources, writing—original draft preparation. T.S.: resources, writing—review and editing. J.R.S.: conceptualization, writing—review and editing.
Disclosures
J.R.S. has received research support from the National Institutes of Health, the Yung Family Foundation, and PCORI. He has received material support from Myriad. He receives royalties from UpToDate, Springer as well as Cambridge. He has provided Continuing Medication Education lectures for the Neuroscience Education Institute, the American Academy of Child & Adolescent Psychiatry, and the American Academy of Pediatrics. He has consulted with FDA, Cerevel, Intracellular Therapeutics, and Otsuka. S.L.S. has received research support from the National Institutes of Health. No other disclosures.
Disclaimer
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Supplementary Material
Supplementary Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.