
Abstract
Introduction:
Escitalopram is an effective and generally well-tolerated antidepressant, but children of parents with bipolar disorder (BD) may be at increased risk for adverse events associated with antidepressants, including increased irritability, restlessness, impulsivity, and manic symptoms. This risk may be influenced by polymorphisms in genes encoding cytochrome P450 enzymes (CYP2C19 or CYP2D6), the serotonin transporter (SLC6A4), and the serotonin receptor 2A subtype (HTR2A). We explored whether gene–drug interactions influence the emergence of adverse events in depressed and/or anxious youth with a family history of BD.
Materials and Methods:
Children and adolescents aged 12–17 years with a first-degree relative with bipolar I disorder were treated with escitalopram and monitored for adverse effects, underwent pharmacogenetic testing, and provided serum escitalopram levels. Emergence of adverse events was determined by study clinicians, and symptoms were tracked using the Treatment-Emergent Activation and Suicidality Assessment Profile (TEASAP) and Pediatric Adverse Events Rating Scale. Clinical Pharmacogenetics Implementation Consortium guidelines were used to determine CYP2C19 and CYP2D6 phenotypes.
Results:
Slower CYP2C19 metabolizers had greater dose-normalized 24-hour area under the curve (AUC0–24; p = 0.025), trough concentrations (Ctrough; p = 0.013), and elimination half-lives (t1/2; p < 0.001). CYP2D6 phenotype was not significantly associated with any pharmacokinetic parameter. Slower CYP2D6 metabolizers had increased TEASAP akathisia (p = 0.015) scores. HTR2A A/A and A/G genotypes were associated with increased TEASAP “self-injury, suicidality, and harm to others” subscale scores (p = 0.017). Escitalopram maximum concentration, AUC0–24, CYP2C19 phenotype, and SLC6A4 genotype were not associated with adverse events.
Conclusions:
CYP2C19 phenotype influences escitalopram pharmacokinetics whereas CYP2D6 phenotype does not. Slower CYP2D6 metabolism was associated with increased akathisia, and HTR2A A/A or A/G genotypes were associated with increased risk of self-harm or harm to others. Larger cohorts are needed to identify associations between genetic test results and antidepressant-associated adverse events.
Trial Registration:
Introduction
Antidepressants are routinely prescribed to treat depression and anxiety in youth (Ramsey et al., 2020), although some youth may experience adverse events after starting antidepressants, including agitation, aggression, suicidal ideation, hypomania, mania, or psychosis (Goldsmith et al., 2011; Joseph et al., 2009). Studies also suggest that when these dysfunctional arousal-like symptoms emerge in youth, they may be more frequent and severe than in adults (Martin et al., 2004; Safer and Zito, 2006; Zuckerman et al., 2007). Bipolar disorder (BD) is a highly heritable condition, and the offspring of parents with BD are more likely to develop depression or anxiety than the offspring of parents who do not have BD (Diler et al., 2011; Hillegers et al., 2005; Singh et al., 2007).
Furthermore, when the children of parents with BD develop depressive or anxiety symptoms, their risk of developing BD increases (Geller et al., 2001). Importantly, youth with a familial risk of BD have an increased risk of developing antidepressant-induced adverse events such as the arousal-like symptoms described above (Baumer et al., 2006; Strawn et al., 2014) and there may be genetic markers that influence the emergence of antidepressant-related adverse events, including manic symptoms.
Selective serotonin reuptake inhibitors (SSRIs) are commonly prescribed to treat depression and anxiety in youth, and these symptoms may represent a prodrome to BD in high-risk youth (Tulisiak et al., 2017). In such youth, it can be difficult to discern whether the emergence of hyperarousal symptoms is attributable to their predisposition to mania or the activating effects of antidepressants (Angal et al., 2019). The potential risk of SSRI-induced manic symptoms in these “at risk” youth with depression or anxiety complicates clinical decision making.
Although several open-label studies have explored alternatives to SSRIs to treat depression or anxiety in high-risk youth, there remains no definitive first-line treatment supported by large randomized controlled trials (DelBello et al., 2007; Findling et al., 2007; Geller et al., 1998). By considering the pharmacogenetics of SSRIs and the emergence of mania-like arousal symptoms, clinicians might be able to guide therapy with genetic testing. If successful, such a strategy would represent the successful application of personalized medicine principles to the treatment of youth at risk for BD.
With these considerations in mind, we treated depressed and/or anxious youth at familial risk for BD with escitalopram, an SSRI approved by the U.S. Food and Drug Administration (FDA) for the treatment of major depressive disorder in adolescents aged 12–17 years and for generalized anxiety disorder in youth aged 7–17 years and older (Emslie et al., 2009; Strawn et al., 2023).
Several genetic factors are relevant to escitalopram pharmacology and the risk of antidepressant-induced adverse events including HTR2A (Garfield et al., 2014; Kato et al., 2006; Murphy et al., 2003; Wilkie et al., 2009), the gene for the serotonin 2A receptor (5-HT2A); SLC6A4 (Chang et al., 2017; Kato and Serretti, 2010; Park et al., 2015), the gene for the serotonin transporter; and genes for cytochrome P450 enzymes involved in escitalopram metabolism, including CYP2C19 (Aldrich et al., 2019; Joas et al., 2022; Veldic et al., 2019) and CYP2D6 (Herrlin et al., 2003; Ozbey et al., 2018; Sindrup et al., 1993; Tsai et al., 2010).
Pharmacogenetic tests have been clinically available for decades. However, studies of their clinical utility, including several randomized clinical trials and combinatorial approaches, in children and adolescents have produced conflicting results (Altar et al., 2013; Amitai et al., 2016; Dong et al., 2016; Laje et al., 2009; Oz et al., 2020; Vande Voort et al., 2022). The Clinical Pharmacogenetics Implementation Consortium (CPIC) recently reviewed evidence for associations between escitalopram and CYP2C19, CYP2D6, HTR2A, and SLC6A4. They concluded there was enough evidence for dose adjustment for CYP2C19 and not CYP2D6, HTR2A, or SLC6A4 (Bousman et al., 2023).
Meanwhile, multiple analyses have suggested a relationship between these pharmacodynamic genes and antidepressant-related adverse events, including the emergence of mania and related symptoms (Chang et al., 2017; Joas et al., 2022). Our analysis builds upon these results by examining a number of genes that are important for escitalopram metabolism and SSRI response, with a focus on adverse events, especially mania-like arousal (or hyperarousal) symptoms that emerge in the context of high-risk youth seeking treatment for depression or anxiety.
In this study, we examined whether pharmacogenetic variants were associated with the risk of escitalopram-related adverse events in depressed and/or anxious youth at familial risk for BD. We hypothesized that CYP450 enzyme metabolizer phenotypes (CYP2C19, CYP2D6) would predict dose-normalized pharmacokinetic parameters and that slower CYP2C19 metabolizers would confer increased risk of escitalopram-related adverse events. We also hypothesized that HTR2A G/G and SLC6A4 S/S genotypes might increase the risk of mania-like symptoms based on observed increases in rates of adverse events among these genotypes in prior studies (Chang et al., 2017; Kato and Serretti, 2010; Kato et al., 2006; Murphy et al., 2003; Park et al., 2015; Wilkie et al., 2009).
Methods
This study was conducted at two sites, the University of Cincinnati and Stanford University, from June 2016 to April 2022, in accordance with the guidelines of the Declaration of Helsinki and was approved by their respective institutional review boards. All legal guardians and youth provided written informed consent and assent, respectively, before engaging in any study procedures.
Participants
We recruited outpatient youth aged 12–17 years who had at least one first-degree relative with bipolar I disorder and clinically significant depression and/or anxiety symptoms (operationalized as a Children's Depression Rating Scale-Revised (Poznanski et al., 1984) score >35 or a Pediatric Anxiety Rating Scale (Anonymous, 2002) score ≥15. Although this analysis was part of a larger randomized placebo-controlled trial, the analysis presented here sought to determine whether gene–drug interactions influence the emergence of adverse effects in response to escitalopram.
Thus, only the escitalopram-treated youth were included. Participants were eligible only if they were antidepressant naïve and had negative urine drug screen and pregnancy test results at the time of enrollment. Participants were excluded if they had any history of BD, autism spectrum disorder, obsessive-compulsive disorder, posttraumatic stress disorder, Tourette's disorder, any psychotic disorder, intellectual disability, any neurological disease, drug or alcohol abuse in the 6 months before study participation, or any unstable medical or psychiatric condition requiring immediate hospitalization.
Participants with comorbid attention-deficit/hyperactivity disorder taking stable doses of stimulants or
Study treatment
Participants were randomized (2:1) to receive treatment with either psychotherapy + escitalopram or psychotherapy + placebo (Honeycutt et al., 2022). All raters and clinicians were blinded to participants' treatment. Escitalopram was initiated at 5 mg daily for 1 week, increased to 10 mg for 1 week, and then titrated by the treating child and adolescent psychiatrists to a target dose of 20 mg/day, with a maximum of 30 mg. Participants' final doses ranged from 10 to 30 mg of escitalopram.
Clinical assessments
Outcomes included (1) a hyperarousal event, operationalized as a two-step worsening on the clinician-rated Clinical Global Impressions-Severity of Activation scale (Bussing et al., 2013) compared with baseline and a Children's Global Assessment Scale (Shaffer et al., 1983) score of <55; (2) maximum scores after baseline obtained from the sum of the severity scores of the 13 arousal-based items on the clinician-reported Pediatric Adverse Events Rating Scale (PAERS) (March et al., 2007); and (3) maximum changes from baseline (or from week 4 in participants with follow-up past week 4) in Treatment-Emergent Activation and Suicidality Assessment Profile (TEASAP) (Bussing et al., 2013) subscale scores, including (1) self-injury, suicidality, and harm to others; (2) mania; (3) akathisia, hyperkinesis, and somatic anxiety; (4) disinhibition and impulsivity; and (5) irritability subscales.
Pharmacogenetic assay
At 8 weeks or upon early termination from the study, participants provided cheek swab samples for genetic testing through a commercial pharmacogenomic test (Myriad Genetics; Mason, Ohio). We used CPIC guidelines (Caudle et al., 2017; Hicks et al., 2015) to assign CYP2C19 and CYP2D6 metabolizer phenotypes based on the alleles detected (CYP2C19: *1, *2,*3, *4, *5, *6, *7, *8, *17; CYP2D6: *1, *2, *2A, *3, *4, *5, *6, *7, *8, *9, *10, *11, *12, *14, *15, *17, *41, gene duplication).
Because the CPIC categories used for analysis are metabolizer phenotypes (e.g., participants with normal rates of CYP2C19 metabolism), and not discrete genotypes (e.g., participants with CYP2C19 *1/*1 alleles), CYP metabolizer groups are referred to throughout this study as phenotypic (rather than genotypic) groups. HTR2A genotype comparisons were made between homozygotes for the −1438G>A (rs6311) allele (G/G) and homozygotes and carriers for −1438A (A/A, A/G), based on the results of prior studies of associations between HTR2A and antidepressant-related adverse events (Kato et al., 2006; Murphy et al., 2003; Wilkie et al., 2009).
SLC6A4 genotype comparisons were based on homozygosity for the “short” risk allele (S/S) versus homozygotes and carriers for the “long” allele (L/L and L/S), as rates of adverse events have tended to be highest among S/S genotyped groups in prior studies (Chang et al., 2017; Kato and Serretti, 2010; Park et al., 2015).
Pharmacokinetic analysis
Participants provided blood samples at 8 weeks or early termination. Serum escitalopram concentrations were measured using ultra-high performance liquid chromatography with tandem mass spectrometry. We used MwPharm (version 3.82; MediWare BV; Groningen, the Netherlands) using the model from Strawn et al. (Poweleit et al., 2023; Strawn et al., 2019) as the Bayesian priors to combine measurements of serum levels with sampling time, escitalopram dose, and adherence data, as well as participants' age, weight, and height, to calculate Bayesian-estimated pharmacokinetic parameters including the 24-hour area under the curve (AUC0–24), maximum concentration (Cmax), trough concentration (Ctrough), and elimination half-life (t1/2) of escitalopram.
We compared these pharmacokinetic parameters across CYP2C19 and CYP2D6 phenotypes, modeled with a 20 mg/day dose to produce dose-normalized pharmacokinetic parameters. Adherence was assessed by participant-completed diaries, pill counts, and clinical reports of adherence assessed during return visits. When full details of dose timing or adherence were unavailable, we assumed that participants took their medication at a consistent time of day commensurate with clinical reports and records of dosing in the 2 weeks preceding the blood draw. We also assumed that the number of doses participants missed was most accurately reflected in pill counts when other records of missed doses (e.g., dose diaries, clinical notes, and self-reports during visits) were incomplete or inconsistent.
Statistical methods
Associations between mania-like hyperarousal and genotypic groups were compared through Fisher's exact test. Associations between dichotomous genotypic groups and continuous outcomes (pharmacokinetic parameters, max PAERS, TEASAP increases) were compared using unpaired t-tests for genotype groups with similar sizes and Mann–Whitney test for groups with dissimilar sizes. Associations between continuous outcomes and genotypic groups with ≥3 groups were analyzed using the method of least squares to fit general linear models for analysis of variance (ANOVA) in the context of an unbalanced distribution of metabolizer phenotypes among participants.
Associations between clinical outcomes and select pharmacokinetic parameters (Cmax, AUC0–24) were analyzed through unpaired t-tests with respect to categorical outcomes (hyperarousal) and through simple linear regression with respect to continuous outcomes (max PAERS and TEASAP increases). Dose-normalized pharmacokinetic data (Cmax, Ctrough, AUC0–24, t1/2) were used to analyze the effects of genes associated with escitalopram metabolism (CYP2C19, CYP2D6), whereas nondose-normalized pharmacokinetic data (Cmax, AUC0–24) were used to analyze associations with clinical outcomes to account for the effects of actual drug exposure on adverse outcomes. Because this is a preliminary study with prespecified genes of interest and hypothesized outcomes, we did not correct for multiple comparisons.
Results
Participants
One hundred and nineteen high-risk youth were enrolled in the study. Among them, 66 participants who provided samples for genetic testing were assigned to the escitalopram group, and 48 participants in this subgroup also provided blood samples for pharmacokinetic analysis. Participants' demographic characteristics are summarized in Table 1.
Participant Demographics and Genotypes
Demographics for escitalopram-treated participants with genetic test results and with blood serum samples used for pharmacokinetic analysis. All 66 were included in analyses of associations between genetic test results and adverse events except as noted above.
Excluded from subsequent CYP2D6 analyses.
IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; RM, rapid metabolizer; SD, standard deviation; UM, ultrarapid metabolizer.
Seven of 66 participants were concurrently taking a stimulant medication (3 took extended-release methylphenidate hydrochloride, 1 took dexmethylphenidate, 1 took lisdexamfetamine, 1 took amphetamine/dextroamphetamine salts, and 1 took extended-release amphetamine/dextroamphetamine salts) and the remaining 59 patients did not take any concurrent psychiatric medications during the study. Hyperarousal events were observed in 14 participants treated with escitalopram, 12 of whom provided cheek swab samples for genetic testing, and 5 of whom provided blood samples for the analysis of serum escitalopram levels.
CYP2C19
CYP2C19 metabolizer phenotypes were almost exclusively rapid metabolizer (RM n = 17), normal metabolizer (NM n = 31), or intermediate metabolizer (IM n = 17), with only one ultrarapid metabolizer (UM) and no poor metabolizers (PMs). We chose to combine CYP2C19 UM and RM phenotypes for all initial analyses. Among the 48 escitalopram-treated participants who provided samples for pharmacokinetic analysis, CYP2C19 metabolizer phenotype had a significant effect on the dose-normalized AUC0–24 (p = 0.025), Ctrough (p = 0.013), and t1/2 (p = 0.0008) of escitalopram (Fig. 1).
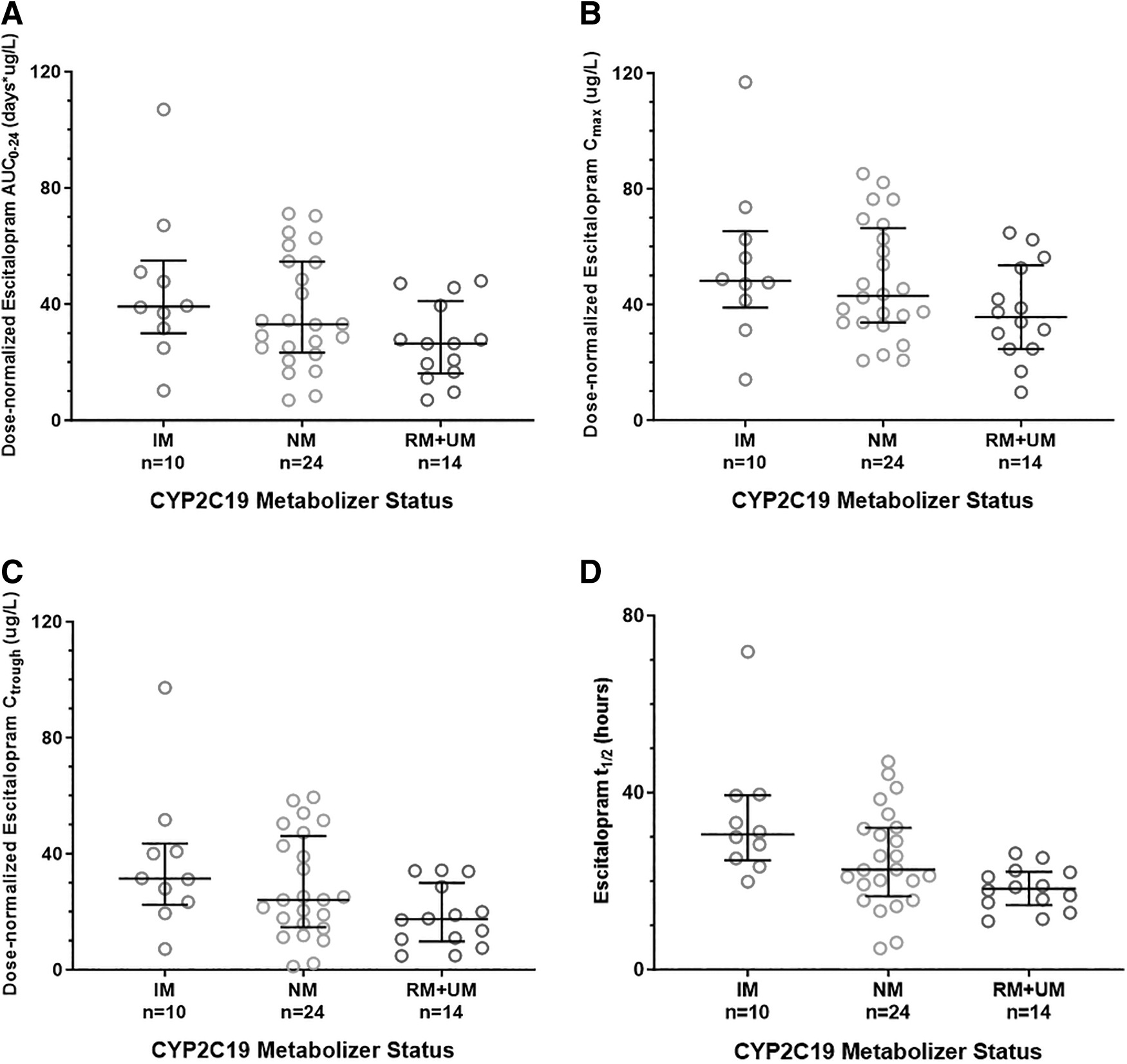
Analysis of associations between CYP2C19 phenotype (including IM, NM, RM, and UM phenotypes) and pharmacokinetic parameters. CYP2C19 phenotype was associated with
Slower CYP2C19 metabolizers also tended to have higher dose-normalized Cmax, though this relationship was not statistically significant (p = 0.057). Slower CYP2C19 metabolizers tended to have greater TEASAP “self-injury, suicidality, and harm to others” subscale scores (F = 2.32, df = 2, p = 0.09), though CYP2C19 phenotype was not associated with increases in other TEASAP subscales scores (p = 0.39–0.92), maximum PAERS score (p = 0.27), or hyperarousal events (p = 0.60; Table 2). There were no significant differences among CYP2C19 phenotypic groups with respect to gender or ancestry.
Analysis of Associations Between CYP Enzymes and Adverse Events
Summary of statistical findings regarding associations between CYP 450 enzyme phenotypes and adverse events. A relationship was observed between the caregiver-reported increases in akathisia-like symptoms and CYP2D6, with slower metabolizers exhibiting greater increases, though this relationship was most pronounced among CYP2D6 PMs that comprised a small (and thus unbalanced) group.
Significant to p < 0.05.
IM, intermediate metabolizer; NM, normal metabolizer; PAERS, Pediatric Adverse Events Rating Scale; PM, poor metabolizer; RM, rapid metabolizer; SD, standard deviation; TEASAP, Treatment-Emergent Activation and Suicidality Assessment Profile; UM, ultrarapid metabolizer.
Comparison of adverse events between an aggregate group of participants with normal or faster than normal CYP2C19 metabolism (NM, RM, or UM) compared to those with slower than normal metabolism (IM) similarly revealed no significant differences in rates of adverse events between aggregate CYP2C19 phenotypic groups. There was no significant association between nondose normalized pharmacokinetic parameters and hyperarousal (Cmax; p = 0.92; AUC0–24; p = 0.74) or change in TEASAP subscales (Cmax; p = 0.16–0.77, R 2 = 0.001–0.054; AUC0–24; p = 0.34–1.00, R 2 = 0.0001–0.055), though participants with lower nondose-normalized AUC0–24 or Cmax tended to exhibit greater PAERS scores (Cmax p = 0.11; AUC0–24 p = 0.11; Table 3).
Analysis of Associations Between Escitalopram Exposure and Adverse Events
Summary of statistical findings regarding associations between pharmacokinetic parameters (i.e., Cmax and AUC0–24) and adverse events. Data were analyzed using linear regressions for continuous measures and an unpaired t-test for hyperarousal. There were no significant associations observed, though clinician-reported max PAERS scores tended to decrease as escitalopram exposure increased.
AUC0–24, 24-hour area under the curve; Cmax, maximum concentration; PAERS, Pediatric Adverse Events Rating Scale; SD, standard deviation; TEASAP, Treatment-Emergent Activation and Suicidality Assessment Profile.
CYP2D6
CYP2D6 metabolizer phenotypes were predominantly normal (NM n = 43) or intermediate (IM n = 15), with few ultrarapid (UM n = 2) or poor (PM n = 4) metabolizers. Two participants were excluded from CYP2D6-related analyses because genetic testing indicated gene duplications without revealing which allele was duplicated, thus precluding a definitive interpretation of CYP2D6 phenotype (Table 1). CYP2D6 phenotype was not associated with any dose-normalized pharmacokinetic parameters (AUC0–24 p = 0.52; Cmax p = 0.48; Ctrough p = 0.56; t1/2 p = 0.50).
Slower CYP2D6 phenotypes were significantly associated with greater increases in TEASAP Akathisia, Hyperkinesis, and Somatic Anxiety subscale (p = 0.015; Fig. 2), but not with other TEASAP subscales (p = 0.14–0.99), maximum PAERS score (p = 0.51), or hyperarousal events (p = 0.55; Table 2). Comparison between an aggregate group with normal or faster than normal metabolism (NM or UM) and an aggregate group with slower than normal CYP2D6 metabolism (IM and PM) revealed that participants with slower than normal CYP2D6 metabolism had significantly greater TEASAP disinhibition and impulsivity subscale scores than those with normal or faster than normal metabolism (mean = 3.0 vs. 1.1, p = 0.02).
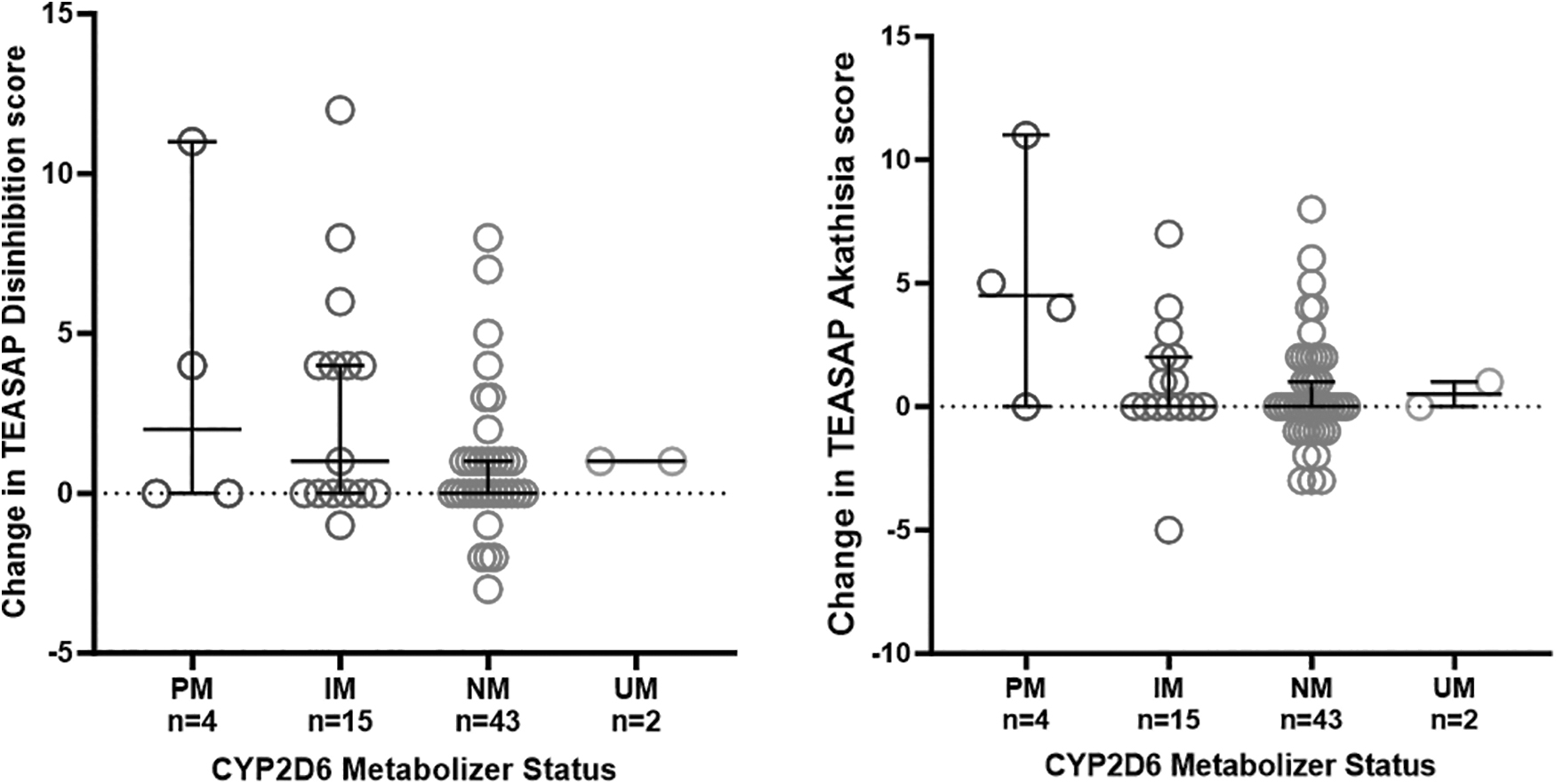
Plots of TEASAP subscale score changes grouped by CYP2D6 phenotype (including PM, IM, NM, and UM phenotypes). Slower CYP2D6 metabolizer phenotypes were associated with greater increases in akathisia subscale scores (p = 0.015). Participants with slower CYP2D6 phenotypes also tended to have greater increases in disinhibition scores, though this finding was not statistically significant (p = 0.14). IM, intermediate metabolizer; NM, normal metabolizer; PM, poor metabolizer; TEASAP, Treatment-Emergent Activation and Suicidality Assessment Profile; UM, ultrarapid metabolizer.
There was also a trend for those with slower than normal 2D6 metabolism to have greater mania and greater akathisia, hyperkinesis, and somatic anxiety TEASAP subscale scores (mania: mean = 2.2 vs. 0.78, p = 0.07; akathisia, hyperkinesis, and somatic anxiety: mean = 1.8 vs. 0.60, p = 0.08). Analysis of demographic characteristics of CYP2D6 phenotypic groups revealed that PM and IM groups were older (p = 0.004) than NM and RM groups, though accounting for age in a covariate model did not meaningfully affect the results reported above. There were no significant differences between CYP2D6 phenotypic groups with respect to gender or ancestry.
HTR2A and SLC6A4
Participants with HTR2A A/A or A/G genotypes had a greater increase in TEASAP “Self-injury, Suicidality, and Harm to Others” subscale scores relative to those with HTR2A G/G genotypes (p = 0.017). We did not have a sufficient number of participants to further analyze differences between participants with A/A versus A/G genotypes. There was no significant difference between these genotype groups with respect to other TEASAP subscales (p = 0.15–0.45), PAERS score (p = 0.39), or the hyperarousal events (p = 0.53; Table 4).
Analysis of Associations Between Pharmacodynamic Genes and Adverse Events
Summary of statistical findings regarding associations between pharmacodynamic genotypes (HTR2A and SLC6A4) and adverse events. Participants with A/A or A/G genotypes had significantly greater increases in the caregiver-reported TEASAP self-injury subscale score. There were no other associations between HTR2A or SLC6A4 genotypes and adverse events.
Significant to p < 0.05.
PAERS, Pediatric Adverse Events Rating Scale; SD, standard deviation; TEASAP, Treatment-Emergent Activation and Suicidality Assessment Profile.
There was no significant difference in clinical outcomes between participants with SLC6A4 L/L or L/S versus S/S genotypes, including TEASAP increases (p = 0.40–0.81), max PAERS score (p = 0.72), or hyperarousal frequency (p = 0.72). There were no significant differences among HTR2A or SLC6A4 genotypic groups with respect to gender or ancestry.
Discussion
Consistent with prior studies (Bousman et al., 2023; Gram et al., 1993; Herrlin et al., 2003; Jeppesen et al., 1996; Sindrup et al., 1993), we found that escitalopram pharmacokinetics were influenced by CYP2C19 but not CYP2D6. However, in contrast to previous studies of youth (Aldrich et al., 2019) and adults (Joas et al., 2022; Veldic et al., 2019), we did not find a relationship between CYP2C19 phenotype and rates of adverse events. Potential reasons for this disparity include the relatively low number of CYP2C19 PMs in our study sample (see limitations below), small sample size, and differences in outcomes measures between studies (e.g., different operationalized definitions of activation/hyperarousal).
Though CYP2C19 phenotype clearly influenced AUC24 and Cmax, and participants with greater AUC24 and Cmax tended to have smaller increases in max PAERS scores, the relationship between escitalopram levels and max PAERS score was not statistically significant and there was no corresponding relationship between CYP2C19 phenotype and max PAERS scores. Though we cannot draw firm conclusions from these results, the observed tendency of participants with higher escitalopram levels to have lower max PAERS scores runs contrary to our initial hypothesis that higher serum escitalopram levels would correlate with increased rates of adverse events.
Our findings also suggest that adverse outcomes of interest were influenced by CYP2D6. Specifically, there were associations between slower CYP2D6 metabolizer phenotypes and increased caregiver reports of akathisia, disinhibition, and manic symptoms. Our findings related to CYP2D6 and adverse events are especially novel and might be related to the effects of an active metabolite of escitalopram, desmethylescitalopram (DCT). Escitalopram is primarily metabolized by CYP2C19, with a minor contribution from CYP2D6.
Escitalopram is metabolized by CYP2C19 to DCT (Bousman et al., 2023), which is then preferentially metabolized by CYP2D6 to didesmethylescitalopram (DDCT) (Gram et al., 1993; Herrlin et al., 2003; Sindrup et al., 1993). DCT is pharmacologically active with a pharmacodynamic profile distinct from escitalopram, whereas the pharmacodynamics of DDCT have yet to be characterized. DCT has a lower affinity for the serotonin transporter relative to escitalopram as well as an increased (though still relatively low) affinity for the norepinephrine transporter (Deupree et al., 2007; Owens et al., 2002; Tatsumi et al., 1997).
Although escitalopram is highly selective for the serotonin transporter, DCT also has low affinity for several additional receptors, including antagonism at the 5-HT2B, 5-HT2C, 5-HT7, and H1 receptors (Deupree et al., 2007). Of note, slower CYP2D6 metabolizers have increased DCT exposure that, in adults with major depressive disorder, may impact treatment response (Ozbey et al., 2018; Tsai et al., 2010). Though the specific pharmacodynamic differences between escitalopram and DCT are of unclear clinical significance, they underscore the potential contribution of DCT to both the side effect profile and the in vivo therapeutic mechanism of escitalopram.
Adverse outcomes of interest were also influenced by HTR2A genotype, as we found greater increases in caregiver reports of high-risk behaviors (i.e., self-harm, suicidality, and harm to others) among those with HTR2A A/A and A/G genotypes. Our findings regarding HTR2A seemed to run contrary to those of prior studies that found greater rates of adverse events among −1438G>A (rs6311) homozygotes (G/G), but a detailed examination reveals a more complex relationship between HTR2A genotype and antidepressant-induced side effects.
Although we found an increase in high-risk behaviors among participants with the −1438A allele (i.e., A/A and A/G genotypes) in response to escitalopram, multiple prior studies have found increased rates of antidepressant-related somatic, but not psychiatric, adverse events among −1438G>A homozygotes (G/G). Homozygosity for the −1438G>A allele (G/G) has been shown to increase the risk of somatic symptoms (e.g., nausea, vomiting, diarrhea, and dizziness) in adult participants treated with paroxetine in multiple studies (Kato et al., 2006; Murphy et al., 2003; Wilkie et al., 2009).
However, one study of escitalopram found higher rates of sexual side effects among adult participants with HTR2A A/A and A/G genotypes (Garfield et al., 2014). As such, different HTR2A genotypes may be associated with different side effect profiles, with a greater risk of side effects such as nausea, vomiting, diarrhea, and dizziness among G/G genotypes, and a greater risk of suicidality, harmful behavior, and sexual side effects among A/A and A/G genotypes.
One potential mechanism for the observed differences in outcomes among HTR2A genotypes is increased signaling through HTR2A among individuals with G/G genotypes, as molecular studies of this genotype suggest greater expression of an extended 5′untranslated region mRNA transcript that may correlate with increased translational efficiency (Smith et al., 2013). As such, individuals with greater HTR2A expression (and thus activity) might be more sensitive to the somatic side effects of SSRIs due to increased expression of HTR2A in vascular and gastrointestinal tissue, and more responsive to treatment with SSRIs due to increased expression of HTR2A in neural pathways underlying symptoms of depression and anxiety.
In contrast to prior studies (Chang et al., 2017; Kato and Serretti, 2010; Park et al., 2015), our analysis did not detect an association between SLC6A4 and clinical outcomes. The S allele of SLC6A4 has been found to increase the likelihood of mania-like arousal symptoms emerging in high-risk youth in association with enhanced activity in, and functional connectivity between, the ventrolateral prefrontal cortex and the right amygdala (Chang et al., 2017).
SLC6A4 has also been implicated in treatment response to SSRIs, as youth who are homozygous for the “short” allele of SLC6A4 (S/S) showed less reduction in depressive symptoms during treatment with citalopram compared with youth who were heterozygous or homozygous for the “long” allele of SLC6A4 (L/S, L/L) (Kronenberg et al., 2007). Reduced treatment response among SLC6A4 S/S genotyped individuals was observed in a separate study of anxious youth treated with escitalopram (Strawn et al., 2020).
Furthermore, high-risk youth with the S/S genotype may have higher baseline anxiety than L/L high-risk youth (Park et al., 2015). Consistent with this, in this study, among the 16 participants with S/S genotypes, 15 (94%) met inclusion criteria for anxiety symptoms compared with 38 of the 50 (76%) participants with L/L or L/S genotypes.
There are several limitations to our study, especially with respect to the association between pharmacokinetic factors and clinical outcomes. First, this analysis may be underpowered to detect the influence of pharmacokinetic factors on the clinical outcomes of escitalopram treatment. Moreover, few participants had extreme CYP2C19 or CYP2D6 phenotypes. Given that increases in rates of adverse events are greatest among PMs (Aldrich et al., 2019; Joas et al., 2022; Veldic et al., 2019), their absence among CYP2C19 phenotypes and low frequency among CYP2D6 phenotypes may have decreased our ability to detect pharmacokinetic gene–drug interactions.
Future studies of the interaction between pharmacokinetic factors and antidepressant-induced adverse events might be designed to recruit participants based on genetic test results at the time of enrollment to ensure adequate distribution and representation of extreme metabolizer phenotypes.
Second, only 9 of the 14 participants who experienced hyperarousal provided blood samples for pharmacokinetic analysis. Therefore, participants who experienced hyperarousal were disproportionately under-represented in our analysis of pharmacokinetic data, thus limiting our ability to detect pharmacokinetic correlates of clinical outcomes. Future studies that seek to detect such interactions should consider gathering samples for pharmacokinetic analysis earlier in the protocol (e.g., after 4 weeks instead of the 8-week threshold used here) to decrease the odds that subjects would experience clinical deterioration that might affect their ability or willingness to provide blood samples.
A third limitation is that we did not correct for multiple comparisons. Our clinical findings should be understood in the context of these multiple comparisons, warranting further study with larger sample sizes to confirm our results. Partially for this reason, this study did not analyze outcomes with respect to anxiety and depressive symptoms as our primary aim was to detect a relationship between participants' genotypes and adverse events. Future analyses will explore these and other data gathered as part of the larger study. Even if firm clinical conclusions cannot be drawn from this analysis, we hope that these results might inform future studies to better identify and characterize genetic correlates of adverse clinical outcomes induced by escitalopram and other antidepressants.
Conclusions
Our study suggests that the pharmacokinetics of escitalopram are influenced by CYP2C19 but not CYP2D6 phenotype. However, CYP2D6 phenotype and HTR2A genotype may influence the risk of adverse effects of escitalopram. Specifically, individuals with slower CYP2D6 phenotypes may be at increased risk of experiencing akathisia or worsening impulsivity. HTR2A genotype likely plays a role in influencing the risk of both somatic and psychiatric adverse events, with individual genotypes associated with varying types of side effects. These findings should be considered in the context of certain limitations in the study design, including small sample size. We hope these results and subsequent studies might improve tolerability and efficacy by informing the use of biomarkers in youth at risk for developing bipolar I disorder.
Footnotes
Acknowledgments
We thank Ethan Poweleit, BS, for his assistance with pharmacokinetic modeling, Zeruneseay Desta, PhD, for measuring the escitalopram concentrations, and Max Tallman, MS, for his assistance with data acquisition and logistical support, and Myriad Genetics for its support with genetic testing. We thank the families who participated in this study and clinical research professionals for their assistance.
Authors' Contributions
Conceptualization of this study was contributed by M.K.S., M.P.D., J.R.S., and D.C.H.; data curation was done by T.J.B. and D.C.H.; formal analysis was carried out by L.B.R., T.J.B., D.C.H., and M.P.D.; funding acquisition was by M.P.D. and M.K.S.; investigation was done by D.C.H., M.P.D., L.B.R., and M.K.S.; methodology was taken care by M.K.S., M.P.D., D.C.H., L.B.R., J.R.S., and J.A.W.; project administration was by M.P.D. and M.K.S.; resources were taken care by M.P.D. and M.K.S.; software was done by T.J.B. and L.B.R.; and supervision of study clinicians was taken care by M.P.D., M.K.S., and L.R.P.
Visualization for Tables 1–4 was by D.C.H. and visualization for Figures 1 and ![]() was by L.B.R.; writing—original draft was taken care by D.C.H., M.P.D., M.K.S., and L.B.R.; writing—review and editing was done by D.C.H., L.B.R., T.J.B., K.M.B., L.R.P., J.R.S., J.A.W., M.P.D., and M.K.S. All authors have read and agreed to the published version of the article.
was by L.B.R.; writing—original draft was taken care by D.C.H., M.P.D., M.K.S., and L.B.R.; writing—review and editing was done by D.C.H., L.B.R., T.J.B., K.M.B., L.R.P., J.R.S., J.A.W., M.P.D., and M.K.S. All authors have read and agreed to the published version of the article.
Disclosures
L.B.R. has received research support from NIH (NICHD). She has received an educational grant and provided consultation to BTG Specialty Pharmaceuticals. J.R.S. has received research support from NIH (National Institute of Mental Health/National Institute of Environmental Health Sciences/NICHD) and AbbVie. He has received material support from Myriad Genetics and royalties from the publication of two texts (Springer). He has served as an author for UpToDate and as an associate editor for Current Psychiatry and has received honoraria from American Academy of Pediatrics, American Academy of Child and Adolescent Psychiatry, and Neuroscience Education Institute.
He receives royalties from UpToDate and Cambridge and has consulted with Cerevel, Otsuka, Intracellular Therapeutics, and the FDA. M.K.S. has received research support from Stanford's Maternal Child Health Research Institute and Stanford's Department of Psychiatry and Behavioral Sciences, National Institute of Mental Health, National Institute of Aging, Patient Centered Outcomes Research Institute (PCORI), Johnson and Johnson, and the Brain and Behavior Research Foundation. She is on the advisory board for Sunovion and Skyland Trail, and is a consultant for Johnson and Johnson, Alkermes, and Neumora.
She has previously consulted for X, the moonshot factory, Alphabet, Inc., and Limbix Health. She receives honoraria from the American Academy of Child and Adolescent Psychiatry, and royalties from American Psychiatric Association Publishing and Thrive Global. M.P.D. has received research support from Alkermes, Allergen, Myriad, Johnson and Johnson, Lundbeck, Otsuka, Pfizer, Sunovion, Supernus, NIMH, and the PCORI. She has conducted consulting for, been on an advisory board of, or received honoraria from Alkermes, Allergan, Assurex, CMEology, Johnson and Johnson, Medscape, Merck, Myriad, Neuronetics Sage, and Sunovion. The remaining authors declare no conflict of interest.