
Abstract
Introduction:
Hyperactive catatonia is often unrecognized in pediatric patients due to its clinical heterogeneity, though it is often seen in children with neurodevelopmental disabilities, especially autism spectrum disorder (ASD). Emerging evidence implicates hyperactive catatonia in more cases of self-injury and aggression in ASD than previously thought.
Objectives:
The study seeks to describe cases of hyperactive catatonia in SYNGAP-1 mutation and examine existing literature for symptomatic overlap between previously-described clinical and behavioral phenotypes of individuals with SYNGAP-1 mutations and catatonia.
Methods:
The study describes two cases of an adolescent and a young adult with SYNGAP-1 mutation and ASD presenting with hyperactive catatonia, which are the first reports of catatonia in individuals known to have a pathogenic variant in SYNGAP-1. A systematic review was undertaken during which 101 articles were screened. 13 articles were then examined for neurological and behavioral features present in participants with SYNGAP-1 mutations which are seen in catatonia.
Results:
The systematic review demonstrates that clinical features suggestive of catatonia are frequently seen among individuals with SYNGAP-1 mutations, including verbal impairment, psychomotor symptoms, aggression, oral aversion, and incontinence. These features were also present in the cases of catatonia in SYNGAP-1 mutations presented here. Both patients showed clinical improvement with use of a long-acting benzodiazepine, and one patient showed benefit from electroconvulsive therapy.
Conclusions:
This symptomatic overlap revealed in the systematic review, including symptoms seen in the reported cases, raises the possibility that diagnoses of catatonia may have been missed in the past in individuals with SYNGAP-1 mutations. Self-injurious behavior and aggression, which are hallmarks of hyperactive catatonia, are commonly part of the behavioral phenotype of SYNGAP-1-related disorders. Clinicians should consider catatonia as a cause of such symptoms in individuals with SYNGAP-1 mutations, as effective treatment can result in significant improvement in safety and quality of life.
Introduction
Catatonia is a heterogeneous disorder with hallmark features, including abnormal psychomotor behavior (Benarous et al., 2018; Walther et al., 2019). The diverse set of clinical presentations possible in catatonia, including increased arousal, immobility, and stupor, make accurate diagnosis challenging (Remberk et al., 2020). Neurodevelopmental disorders and genetic syndromes commonly present in cases of pediatric catatonia (Hauptman et al., 2023; Raffin et al., 2018; Smith et al., 2024b), including ∼10% of those with a diagnosis of autism spectrum disorder (ASD) in a recent meta-analysis (Vaquerizo-Serrano et al., 2022). In 2009, mutations in the SYNGAP-1 gene were first identified in nonsyndromic intellectual disabilities (Hamdan et al., 2009). SYNGAP-1 is a gene on chromosome 6p21.32. It encodes a protein that activates synaptic RAS-GTPase and helps form the postsynaptic density, which is a complex of proteins associated with N-methyl-D-aspartate (NMDA) receptors (Chen et al., 1998). It is expressed mainly on excitatory neurons and is known to play a role in the modulation of synaptic plasticity (Kim et al., 1998).
Despite catatonia being reported as a comorbidity in other neurodevelopmental disorders, (Hauptman et al., 2023; Raffin et al., 2018; Smith et al., 2024a; Termini et al., 2023; Vaquerizo-Serrano et al., 2022), there are no previously reported cases of catatonia in individuals of SYNGAP-1-related disorders. In individuals with ASD and pediatric populations, catatonia most often presents with externalizing symptoms, including aggression, self-injury, hyperactivity, loss of previously acquired abilities, reduced oral intake, emotional dysregulation, and profound oppositionality or negativism (Benarous et al., 2018; Smith et al., 2024b; Vaquerizo-Serrano et al., 2022). Many of these symptoms are commonly reported in the SYNGAP-1 literature (Vlaskamp et al., 2019; Wright et al., 2022). Previous research in SYNGAP-1 has described behaviors, including oppositionality, aggression, self-injury, and affective instability (Vlaskamp et al., 2019); all are symptoms consistent with catatonia occurring in autism broadly (Vaquerizo-Serrano et al., 2022), and ASD is commonly diagnosed in individuals with SYNGAP-1 mutations (Wright et al., 2022). There is growing awareness of catatonia in neurodevelopmental disorders associated with specific genetic conditions, including SHANK3 mutations (Phelan McDermid Syndrome) (Dhossche et al., 2023), Trisomy 21 (Smith et al., 2024a), and 22q11.2 syndrome (Termini et al., 2023). The association between SHANK3 mutation and catatonia is especially relevant given that both this condition and SYNGAP-1 mutation are among the most common monogenic neurodevelopmental syndromes associated with ASD (Satterstrom et al., 2020). In addition, both implicated genes are involved in regulation of excitatory neurotransmission (Monteiro and Feng, 2017; Wang et al., 2013), possibly increasing the risk of catatonia (Smith et al., 2023a). A recent publication by Raffin and colleagues reports the largest sample size of children who experienced catatonia and received genetic testing (N = 51), reporting 19 of 51 patients in their cohort (21.3%) with some abnormality on genetic testing (Raffin et al., 2018).
Untreated catatonia is associated with high rates of morbidity and mortality but treatment with benzodiazepines, electroconvulsive therapy (ECT), and other pharmacologic agents can be highly effective (Rogers et al., 2023). Despite these encouraging reports of treatment, differentiation of catatonia symptoms from ASD and SYNGAP-1 mutation-related differences is challenging due to symptomatic overlap. Psychomotor symptoms associated with catatonia, including stereotypies, are part of the core diagnostic criteria for ASD in the Diagnostic and Statistical Manual of Mental Disorders, 5th edition (2013). Significant symptom overlap outside of psychomotor symptoms also exists, including mutism, negativism, and abnormal speech (Carroll et al., 2008). Excited catatonia may be underrecognized, especially in populations with ASD who may exhibit psychomotor symptoms prior to the onset of catatonia (Vaquerizo-Serrano et al., 2022) and may be misattributed to oppositionality or defiance rather than a treatable condition (Hickox et al., 2024). This may explain the significant delay in catatonia diagnosis compared with neurotypical individuals (Zappia et al., 2024). Physical examination findings which can aid the clinician in the differentiation of catatonia from underlying ASD symptoms include posturing, waxy flexibility, ambitendency, catalepsy, new urinary incontinence, acrocyanosis, and schizophasia (Benarous et al., 2018, 2016; Carroll et al., 2008; Vaquerizo-Serrano et al., 2022).
These findings suggest catatonia may be an identifiable and treatable cause of “secondary regression” (a global loss in level of functioning, including motor, speech, social, self-care, or academic skills with onset later than the “early regression” preceding or concurrent with ASD diagnosis) (De Stefano et al., 2023) as well as aggression in SYNGAP-1. To further investigate this possibility, we report two cases of catatonia occurring in individuals with comorbid SYNGAP-1-related intellectual disability. Consent to publish these reports was obtained by the guardians of both patients. To address the phenotypic overlap of catatonic symptoms in SYNGAP-1, we have also conducted a systematic review of the literature in an attempt to recognize catatonia-spectrum symptoms reported in the literature that were not identified as such.
Case 1
A 19-year-old female with a history of monoallelic mutation of the SYNGAP-1 gene, intellectual disability, epilepsy, ASD, and chronic pain presented to the psychiatry clinic for neurodevelopmental disorders for consultation. A summary of case 1 can be found in Table 1.
Summary of Case 1
BFCRS, Bush Francis Catatonia Rating Scale; BID, two times per day; KCE, Kanner Catatonia Examination; ECT, electroconvulsive therapy; KCRS, Kanner Catatonia Rating Scale Severity Measure; TID, three times per day; QAM, every morning; QHS, nightly; QPM, every afternoon.
ASD with intellectual disability due to SYNGAP-1 mutation was diagnosed in childhood. The patient experienced a period of developmental regression with increased behavioral symptoms around age 14. She intermittently experienced behavioral dysregulation with aggression or self-injurious behavior thereafter. Her mother reported discrete episodes over the preceding several years of acutely altered behavior, including little to no oral intake, reduced speech, episodic nudism, increased self-injurious behavior, increased stereotypies, sleep dysregulation, impulsivity, increased aggression, and psychomotor excitement. These episodes were typically constant for several weeks and would then resolve. A total of two selective serotonin reuptake inhibitors, one stimulant, three antipsychotics, and one alpha-2 receptor antagonist had been previously prescribed without significant improvement in aggression or other behavioral symptoms. Magnetic resonance imaging (MRI) of the brain at age 15 that demonstrated mild diffuse volume loss but no other abnormalities were noted. Laboratory workup including complete blood count, complete metabolic panel, thyroid studies, and antinuclear antibody screen completed during the year of presentation did not show any notable abnormalities.
The patient was evaluated in the neurodevelopmental psychiatry clinic for consultation due to these cyclical episodes of aggression. While there was initial concern for catatonia contributing to these waxing and waning episodes of aggressive behavior, the patient was without significant aggression at that time and no new medications were initiated. The patient later experienced an acute regressive episode with significantly increased impulsivity, aggression, and self-harming behaviors along with decreased oral intake and new mannerisms. She was briefly hospitalized without any medication changes and then presented to clinic for follow-up. The patient’s exam demonstrated notable findings of impulsivity, rigidity, combativeness, new mannerisms, and grimacing. She was diagnosed with catatonia at that time, also exhibiting acute onset of new impairments in verbal abilities, decreased oral intake, and nudism. Clonazepam was initiated and increased over the course of several weeks with improvement of oral intake and no evidence of sedation, but other symptoms remained persistent.
Over the next year, clonazepam, memantine, and valproate were utilized in the treatment of catatonia. The patient experienced significant improvement in signs and symptoms of catatonia but with many symptoms remaining. She remained free from significant sedation or ataxia, which are side effects of concern with benzodiazepines at this dose. Over the next few months, catatonia symptoms worsened with recurrent episodes of aggression which were not sufficiently managed with medication, so ECT was initiated. Once satisfactory motor and electroencephalographic response was achieved with bitemporal stimulation, treatment settings were maintained at pulse width 0.5 milliseconds, pulse frequency 70 Hertz, stimulus duration 8 seconds, current 0.9 Amperes, and total charge 501.3 millicoulombs. After 11 treatments, there had been minimal improvement in catatonia symptoms. After a total of 21 ECT treatments, the patient had experienced significant improvement, including decreased frequency and intensity of aggression as well as increased verbal output.
Case 2
A 16-year-old female with a history of ASD level 3 with accompanying intellectual and language impairment with ongoing workup for developmental regressions and behavioral changes presented to the emergency department (ED). A summary of case 2 can be found in Table 2.
Summary of Case 2
BFCRS score is estimated based on chart review.
BFCRS, Bush Francis Catatonia Rating Scale; BID, two times per day; EEG, electroencephalogram; ER, emergency room; KCE, Kanner Catatonia Examination; KCRS, Kanner Catatonia Rating Scale Severity Measure; MRI, magnetic resonance imaging; TID, three times per day; QAM, every morning; QHS, nightly; QPM, every afternoon; Q1H, every hour; Q4H, every 4 hours; PRN, as needed.
Psychiatry was initially consulted for hallucinations and worsening aggression. The patient’s parents reported unprovoked aggression and hallucinations which had gradually increased in frequency and duration over the past 2 years. The patient experienced regression in physical and cognitive abilities, severe insomnia, and autonomic changes when agitated (i.e., flushing, mydriasis, and diaphoresis). Previous workup was notable for unremarkable brain MRI, lumbar puncture, chromosomal microarray, Fragile X testing, and single gene MECP2 sequencing. Whole exome sequencing (WES) was ordered, but aggression had prevented blood collection. During electroencephalogram (EEG) lead placement, the patient’s agitation responded well to intranasal midazolam. The EEG showed minor slowing and no seizure-like activity. She was started on scheduled and as-needed lorazepam, which resulted in improvement in agitation and aggressive behaviors. Laboratory studies were collected during this hospitalization, including vitamin B1 and B12, autoimmune encephalitis panel, and central nervous system’s demyelinating disease panel, which did not show any abnormalities. WES with mitochondrial analysis revealed a SYNGAP-1 pathogenic variant after discharge.
Four days after discharge, the patient presented with aggression in the emergency department. Further history obtained from the patient’s mother appeared consistent with agitated catatonia (i.e., history of staring, repetitive phrases, repetitive movements, impulsivity, grasp reflex, rigidity, withdrawal, and combative behavior). After starting scheduled lorazepam, the patient’s mother reported a reduction of aggression and significant increase in food intake. After discharge, the patient received psychiatric care on an outpatient basis. Due to relapsing symptoms with episodes of worsening aggression, the patient was hospitalized on two further occasions over the course of the following weeks, and catatonia was again identified as the major contributing factor. Despite initial benefit with lorazepam, there were concerns about breakthrough catatonia symptoms despite escalation of the dose. Multiple psychiatric medications, including valproate, failed to address breakthrough catatonia symptoms. The patient had significant improvement of catatonia after addition of memantine and high-dose diazepam and ultimately did not require ECT.
Literature Review
Methods
We conducted a Preferred Reporting Items for Systematic Reviews and Meta-Analyses systematic review of peer-reviewed international literature (Fig. 1) published with no lower limit on date of publication until May 3, 2023. The aim of our review was to examine the clinical features exhibited by individuals with SYNGAP-1 mutations. We searched the PubMed electronic database using the following terms: “SYNGAP” or “SYNGAP1” or “SYNGAP1 mutation” or “SYNGAP1 epilepsy” or “SYNGAP1 gene mutation” and “behaviors” or “catatonia” or “catatonic schizophrenia” or “aggression” or “stupor” or “psychomotor agitation.” Title review was conducted with ASreview, an open-source machine learning framework for efficient and transparent systematic reviews (van de Schoot et al., 2021). A list of signs and symptoms of interest was generated based upon the BFCRS (Bush Francis Catatonia Rating Scale), KCRS (Kanner Catatonia Rating Scale Severity Measure), and Pediatric Catatonia Rating Scale (PCRS) (Benarous et al., 2016). Each article was analyzed by the authors, and the presence or absence of these signs and symptoms in case descriptions or tables describing the number of participants with given symptoms was recorded.
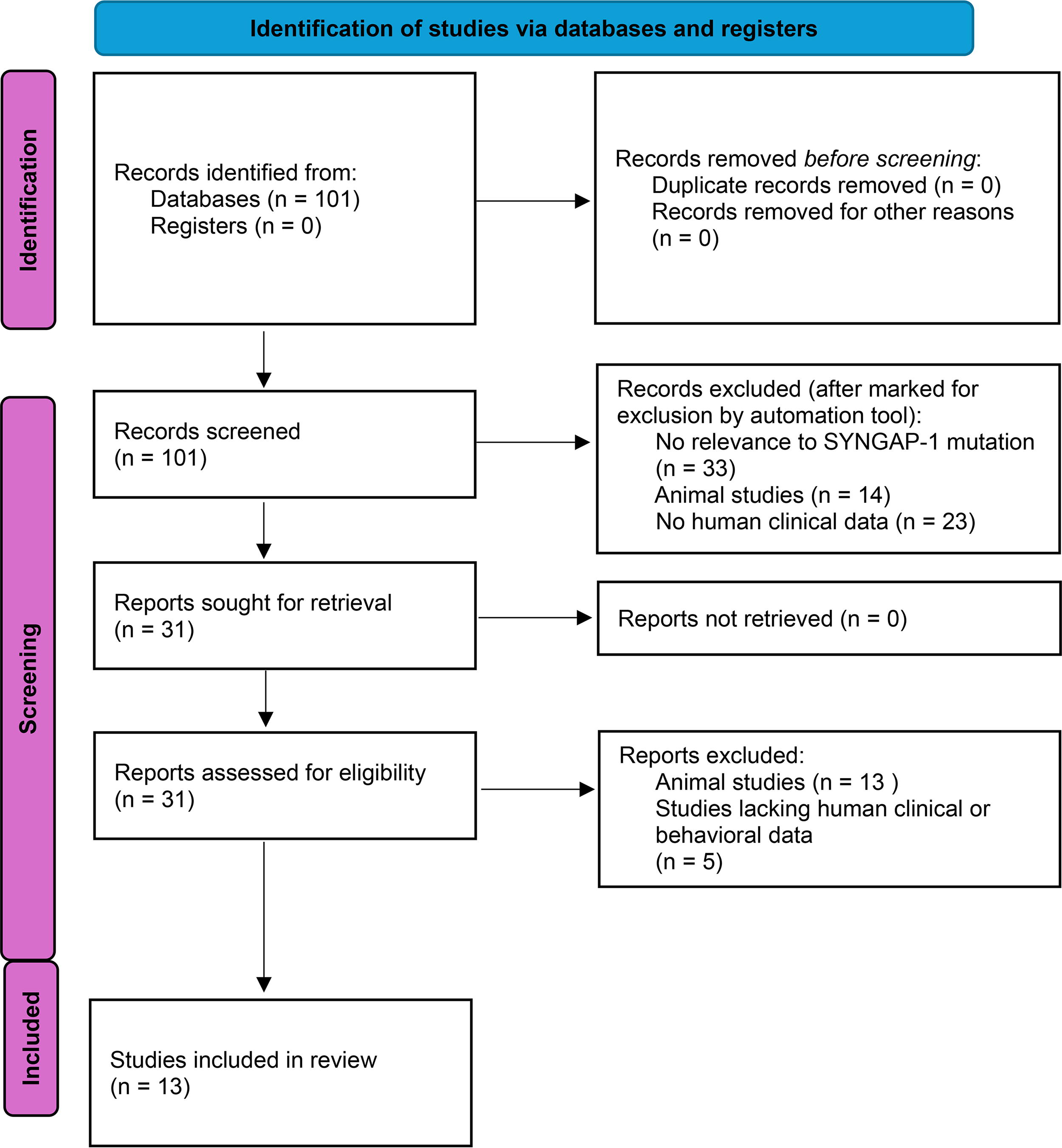
PRISMA flow diagram. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Results
An initial search revealed 101 articles; all were identified from PubMed. No duplicates were removed. A total of 70 articles were removed by initial screening using ASreview (van de Schoot et al., 2021), with reviewers (I.B. and J.R.S.) confirming exclusion by reviewing article titles. These 70 articles included 33 studies without relevance to SYNGAP-1 mutation, 14 studies in animals, and 23 other articles without clinical or behavioral data from human subjects. Abstracts were reviewed for 31 articles, with 13 animal studies and 5 articles lacking clinical data from human subjects removed. A total of 13 studies were identified meeting criteria for inclusion in the review. All articles reviewed are contained in supplementary data.
The studies included a variety of methodologies, including case reports and case series as well as qualitative data collection. Nine of the 13 articles utilized a clinical exam, and 8/13 utilized history obtained from parents or caregivers. Data from a total of 175 individuals between ages 18 months and 33 years are captured in these studies, including 88 males and 87 females. The majority were children or adolescents <18 years of age at the time of assessment. While diagnoses other than SYNGAP-1 mutation were not reported for all individuals in the sample, 72 (42%) had been diagnosed with ASD, 135 (77%) with intellectual disability, and 119 (68%) with epilepsy. The articles were reviewed for neurological and behavioral features present in participants with SYNGAP-1 mutations, and results are presented in Table 3. Motor and behavioral features shared between the participant phenotypes and signs and symptoms of catatonia as outlined by the BFCRS and KCRS are described below.
Literature Review and Shared Clinical Findings with Catatonia
Median age provided rather than mean.
ND, no data for the finding; X, present in sample but no number of participants provided.
Verbal impairment
Many patients with SYNGAP-1 mutations are nonverbal or have verbal impairments (Mignot et al., 2016; Vlaskamp et al., 2019). This was true for many of the participants in the articles reviewed here, with these impairments noted in 10/13 of the studies. At least 27% of the individuals in these studies were described as nonverbal, and 19% were noted to have a verbal impairment. As mutism is a diagnostic criterion for catatonia (Benarous et al., 2016; Bush et al., 1996; Carroll et al., 2008), it is important to differentiate chronic verbal status from catatonic decline in the acute setting in a patient with SYNGAP-1. The course of verbal impairment in SYNGAP-1 can be variable, however, with some patients experiencing regression in verbal skills after learning some words early in life and others experiencing improvement in communication with better control of seizures (Mignot et al., 2016).
Psychomotor behavior
Hyperactivity and repetitive behaviors are also common in these patients (Wright et al., 2022) and can be difficult to disentangle from one another clinically, especially when impulsivity and aggression are also present. Psychomotor behavioral symptoms such as these are core to catatonia, including disturbances in posture, gaze, gesture, and speed and fluidity of movement (Heckers and Walther, 2021). In the articles reviewed, patients exhibited a variety of such signs. Stereotypies, including involuntary repetitive movements of various kinds, were mentioned in 5/13 studies, including at least 12% of the individuals studied.
Rigidity may be an uncommon finding in patients with SYNGAP-1 mutations without catatonia, as many patients experience hypotonia at baseline (Vlaskamp et al., 2019). Hypotonia was commonly reported in the studies reviewed, including in 7/13 studies and at least 38% of individuals studied. While only muscle rigidity appears on the BFCRS (Bush et al., 1996), which is a commonly used measure across all populations, flaccidity is included in the KCRS for neurodevelopmental disorders (Carroll et al., 2008). The hypotonia found in SYNGAP-1-related disorders may be confounding or evidence of catatonia depending on the patient’s baseline muscle tone.
One notable symptom noted in the literature is eyelid myoclonia, a type of seizure involving eyelid fluttering with eyes partially open or closed. This has been described in those with SYNGAP-1 mutations (von Stülpnagel et al., 2019) and could be misinterpreted as abnormal psychomotor behavior or negativism related to catatonia (Vlaskamp et al., 2019). The high prevalence of epilepsy in this population warrants caution in the interpretation of such movements.
Emotional dysregulation and aggression
Emotional dysregulation and aggression are common reasons for ED visits for autistic children and adolescents (Wolpert et al., 2023); these symptoms are also frequent reasons for referral to psychiatry. These are common features among those with SYNGAP-1 mutations. Emotional dysregulation was mentioned in 6/13 studies, including at least 30% of individuals. Nine out of 13 studies described aggression, including at least 14% of individuals. Self-injurious behavior can be seen in individuals with ASD, resulting from emotional dysregulation or abnormal psychomotor behavior such as repetitive mannerisms or stereotypies and is another diagnostic feature of catatonia (Benarous et al., 2016; Bush et al., 1996; Carroll et al., 2008). These behaviors were present in 6/13 studies, and at least 6% of patients with SYNGAP-1 were noted to exhibit this behavior. It is well known that operant factors (factors that act as antecedents or reinforcers of behavior) often contribute to aggressive and self-injurious behavior in ASD (Edelson, 2022). These factors are likely still at play in individuals with catatonia. It is vital to identify these factors, delineate them from catatonia symptoms, and intervene on antecedents and reinforcers of behaviors. When catatonia is determined to be a specific cause of aggression and self-injury, rapid treatment may result in significant improvements in safety for patients and their families.
Oral aversion
Poor oral intake and refusal of food and fluids is another feature of catatonia (Bush et al., 1996; Carroll et al., 2008). In the studies reviewed, 4/13 mentioned either oral aversion, poor intake of food or fluids by mouth, or difficulty with chewing or swallowing, including at least 18% of individuals. Oral aversion was a common finding in the studies led by Wright (Wright et al., 2022) and Vlaskamp (Vlaskamp et al., 2019), with a small minority of patients even requiring feeding tubes. Some patients are known to have seizures triggered by eating, which could contribute to avoidance of oral intake (Vlaskamp et al., 2019). There was, however, a minority of patients in the study by Vlaskamp et al. who experienced hyperphagia with ingestion of inedible objects. One case report notes intermittent episodic periods of total refusal of food beginning at age 3, which resolved with periods of normal oral intake between these episodes (Prchalova et al., 2017, p. 31).
Incontinence
Bladder and bowel incontinence is a feature of catatonia described in the KCRS and PCRS (Benarous et al., 2016; Carroll et al., 2008). Toileting problems were mentioned in 3/13 studies, and at least 10% of individuals exhibited this feature. Worsening difficulty with bowel or bladder control should be quickly recognized in those with SYNGAP-1 mutation and a full examination to rule out catatonia as a potential cause in addition to other medical causes.
Ataxia and gait abnormalities
Ataxia and gait abnormalities are common in patients with SYNGAP-1 mutation, with some studies demonstrating this symptom in a majority of patients (Vlaskamp et al., 2019; Wright et al., 2022). Gait abnormalities were seen in 7/13 studies, including at least 35% of individuals. This is relevant to the treatment of catatonia given the potential side effect of ataxia with use of benzodiazepines. Still, it is not uncommon for patients with SYNGAP-1-related epilepsy be prescribed a long-acting benzodiazepine (LABZD) such as clobazam (Vlaskamp et al., 2019; Wright et al., 2022), so this could be easily implemented into the treatment regimen for some patients.
Discussion
Considering catatonia in SYNGAP-1 mutation
This literature review did not reveal any previously reported cases of catatonia diagnosed in individuals with SYNGAP-1 mutations, making these two cases likely the first described. The studies reviewed here include phenotypic data from a variety of study designs into a single literature review, demonstrating the commonality of several clinical features in individuals with SYNGAP-1 mutations from sources, including clinical examination, caregiver reports, and documented medical histories. Review of the articles demonstrated commonalities between clinical phenotypes in SYNGAP-1 mutation and signs and symptoms of catatonia described in multiple catatonia rating scales, though the symptoms were not conceptualized as being related to catatonia in the articles themselves. The identification of these overlapping signs and symptoms raises the possibility that catatonia may be the underlying cause of such symptoms with SYNGAP-1 mutations. The diagnosis of catatonia may have been missed in the past due to the same factors making diagnosis of catatonia in ASD generally challenging, namely the overlap of baseline features with those seen in catatonia (Vaquerizo-Serrano et al., 2022). Further research into the prevalence and management of catatonia in this population is needed to inform clinical practice. To help guide this future work, an exploration of possible mechanisms, treatment implications, and the limitations of the review follows.
Excitatory–inhibitory imbalance
The connection between ASD and catatonia has yet to be explained on a neurobiological level. One theory suggests excitatory–inhibitory (E/I) imbalance due to a deficiency of parvalbumin resulting in impairment in long-term plasticity may result in cortical hyperexcitability in ASD (Rubenstein and Merzenich, 2003), along with GABAergic dysfunction that has also been demonstrated in catatonia (Wachtel and Dhossche, 2010; Walther et al., 2019). In support of this hypothesis, recent diagnostic work in transcranial magnetic stimulation of intellectually capable persons with autism has reported enhanced cortical modulation, indicative of cortical hyperexcitability (Jannati et al., 2022). Furthermore, recent preliminary magnetoencephalographic research has reported greater E/I imbalance in biologically male autistic individuals with below-average IQ compared with typically developing or autistic persons with average to above-average IQ (Manyukhina et al., 2022), indicative of a correlation between the degree of E/I imbalance and symptomology.
If E/I imbalance is truly a risk factor for catatonia, then SYNGAP-1 mutations may pose an especially high risk, as the E/I imbalance has mechanistic support in SYNGAP-1-mutated animals. The SYNGAP protein normally acts as part of a signaling pathway that suppresses protein synthesis necessary for normal insertion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors on excitatory axons, resulting in abnormally high synaptic strength when mutated (Wang et al., 2013). In fact, mouse models of ASD often demonstrate abnormally high synaptic strength (Bateup et al., 2011; Santini et al., 2013), and mice heterozygous for SYNGAP have demonstrated similar behavioral and cognitive findings to the ASD models (Clement et al., 2012). It has been hypothesized that early maturation of dendritic spines seen in SYNGAP-1 −/+ mice results in E/I imbalance by way of elevated excitatory neurotransmission (Jeyabalan and Clement, 2016). Introduction of a pharmacologic agent meant to restore the functioning of GABAergic neurons via normalization of intracellular chloride concentrations normalized E/I imbalance via restored synaptic plasticity and improved behavioral deficits in mice (Verma et al., 2022), suggesting a link between GABAergic inhibitory transmission and the behavioral phenotypes seen in SYNGAP-1 mutation.
Treatment implications
Lorazepam is frequently used as the treatment of choice for catatonia, though LABZD treatment and ECT have been reported as efficacious for catatonia in autism and intellectual disability (Smith et al., 2023b, 2023a; Wachtel and Dhossche, 2010). Recently, the tolerability of lorazepam for catatonia occurring in ASD has been called into question (Park et al., 2020; Smith et al., 2022; Vaquerizo-Serrano et al., 2022; Wachtel, 2019). The relative inefficacy of lorazepam compared with diazepam seen in case 2 is consistent with these findings. These cases provide further support to the efficacy of LABZD treatment for catatonia in individuals with neurodevelopmental disorders. Though known to be effective for the treatment of catatonia, use of ECT in the United States is limited due to a lack of provider availability, stigma, and underutilization, especially in rural settings (Espinoza and Kellner, 2022; Ong et al., 2023). ECT was effective in case 1 to treat symptoms of catatonia that were insufficiently controlled with LABZD treatment.
As catatonia in neurodevelopmental disorders becomes better recognized, establishing standards of care for treatment is increasingly important. The British Association of Psychopharmacology has recently recommended clinical vigilance for catatonia in ASD, requiring an acute change from baseline for diagnosis (Rogers et al., 2023). They also recommend psychological interventions and/or lorazepam for cases of mild catatonia. In moderate to severe cases of catatonia, benzodiazepines in escalating doses and/or bilateral ECT are recommended. It is notable that in case 1 presented here, 21 ECT treatments were required for catatonia symptoms to reach a negligible level. This is in line with emerging reports of extended courses of ECT treatment for patients diagnosed with ASD, including those with catatonia (Ghaziuddin et al., 2023).
Recognition of the benefits of outpatient assessment and treatment of catatonia in neurodevelopmental disorders is also growing, and some recommendations based upon clinical experience have recently been developed (Ferrafiat et al., 2024). As in these cases, establishing a diagnosis of catatonia in neurodevelopmental disorders can be challenging, though improved by assessment of a personalized catatonia score at baseline on a standardized scale with longitudinal follow-up assessing for catatonic deterioration (Hauptman et al., 2023). These cases provide further evidence that catatonia in neurodevelopmental disorders, including SYNGAP-1 mutation, can be diagnosed and treated on an outpatient basis using treatments including benzodiazepines and ECT.
Limitations
The review has several important limitations. Some studies reported inclusion of cases in their sample who had been previously reported in the literature (11 individuals in Vlaskamp’s description of 57 patients) (Vlaskamp et al., 2019). If some of these cases are reported in other studies included in this review, it could lead to an overestimation of the prevalence of the clinical features present in those individuals. Due to the variability in methodology and reporting of results for the studies included in the literature review, it was difficult to estimate the prevalence of the clinical signs and symptoms described. It was also not possible to determine whether the symptoms described in these articles occurred in discrete episodes or were present at baseline for individual patients.
The studies reviewed have a variety of sources for participants, including existing databases, focused on neurodevelopmental disabilities as well as clinical service outreach and social media advertisements. Each of these samples has unique features that could affect the phenotypic data. For example, there are some individuals reported in the data who are described as having borderline intellectual function, while other studies recruited only those with SYNGAP-1-related intellectual disability. Differing levels of intellectual function may have implications for behavioral phenotypes. Some of the articles focused on description of specific findings, such as seizures (von Stülpnagel et al., 2019), gait abnormalities (Layne et al., 2022), or sensory processing phenotypes (Lyons-Warren et al., 2022). This likely leads to exclusion of relevant findings that are described in other studies and may lead to an underestimate of the presence of those findings as these individuals were included in calculations of prevalence. One study described the behavioral phenotype of only one participant out of eight individuals examined in the study (von Stülpnagel et al., 2019), so only this individual’s data were included in this review.
Conclusions
Catatonia is becoming better recognized as a treatable cause of behavioral regression as well as externalizing symptoms, including aggression, impulsivity, and self-injury, in ASD. These two cases of catatonia in patients with SYNGAP-1 mutations and ASD were recognized by the presence of such features. A review of relevant literature demonstrates significant phenotypic overlap between symptoms experienced by individuals with SYNGAP-1 mutations and catatonia. When considered alongside these two novel cases of catatonia, this overlap raises the possibility that diagnoses of catatonia may have been missed in the past in individuals with SYNGAP-1 mutations.
Clinical Significance
Clinicians should evaluate for catatonic symptoms in patients with a history of SYNGAP-1-related intellectual disability or ASD with regression in skills or aggressive behavior using standardized tools, including the BFCRS, KCRS, and PCRS. Special attention should be given to any history of acute changes in verbal abilities, psychomotor symptoms, and oral intake. LABZDs and memantine were effectively employed in the treatment of catatonia in both cases, while ECT was effective for refractory symptoms in one case. Future research should continue to investigate the potential connection between catatonic features and SYNGAP-1 mutation as well as the role of E/I imbalance in catatonia occurring in the setting of neurodevelopmental disorders.
Footnotes
Acknowledgments
We thank the patients and their families for contributing their medical histories to this research. We thank the National Institute of Child and Human Development for funding support.
Authors’ Contributions
I.B.- conceptualization, methodology, investigation, data curation, formal analysis, writing- original draft, writing- review and editing; A.C.- investigation, writing- original draft, writing- review and editing; N.W.- investigation, writing- original draft, writing- review and editing; G.O.- investigation, writing- original draft, writing- review and editing; D.N.- investigation, writing- original draft, writing- review and editing, supervision; J.R.S.- conceptualization, methodology, investigation, data curation, writing- original draft, writing- review and editing, supervision, project administration, software.
Disclosure
J.R.S. receives funding from Roche, Axial Therapeutics, and the National Institute of Child and Human Development. All other authors have no conflicts of interest.
Supplementary Material
Supplementary Data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.