
Abstract
Background:
Antidepressant medications, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), are commonly used to treat depressive, anxiety, and obsessive-compulsive disorders in youth. Yet, data on discontinuing these medications, withdrawal symptoms, and strategies to switch between them are limited.
Methods:
We searched PubMed and ClinicalTrials.gov through June 1, 2024, to identify randomized controlled trials assessing antidepressant discontinuation in youth. We summarized pediatric pharmacokinetic data to inform tapering and cross-titration strategies for antidepressants and synthesized these data with reports of antidepressant withdrawal.
Results:
Our search identified 528 published articles, of which 28 were included. In addition, 19 records were obtained through other methods, with 14 included. The corpus of records included 13 randomized, double-blind, placebo-controlled trials (3026 patients), including SSRIs (K = 10), SNRIs (K = 4), and TCAs (K = 1), ranging from 4 to 35 weeks. Deprescribing antidepressants requires considering clinical status, treatment response, and, in cross-titration cases, the pharmacokinetics and pharmacodynamics of both medications. Antidepressant withdrawal symptoms are related to the pharmacokinetics of the medication, which vary across antidepressants and may include irritability, palpitations, anxiety, nausea, sweating, headaches, insomnia, paresthesia, and dizziness. These symptoms putatively involve changes in serotonin transporter expression and receptor sensitivity, impacting the serotonin, dopamine, and norepinephrine pathways.
Conclusions:
Although approaches to deprescribing antidepressants in pediatric patients are frequently empirically guided, accumulating data related to the course of relapse and withdrawal symptoms, as well as the pharmacokinetic and pharmacodynamic properties of medications, should inform these approaches. Recommendations within this review support data-informed discussions of deprescribing—including when and how—that are critically important in the clinician–family–patient relationship.
Introduction
Deprescribing is part and parcel of any psychopharmacologist’s practice, namely stopping medications when they do not work, are not tolerated, or when a patient has reached and sustained remission. The contemporary practice of psychopharmacology recognizes that prescribers are also deprescribers, and patients are deprescribers when they are nonadherent, encounter medication access problems, etc. Pediatric psychopharmacology requires clinicians to know how to stop medications, including those that are particularly problematic when discontinuing and are associated with withdrawal symptoms, and what to expect when these medications are discontinued. Yet, clinicians treating children and adolescents have limited data to guide them in deciding when and how to deprescribe, particularly antidepressant medications, which are among the most common psychopharmacologic treatments used in youth.
Among the most used psychotropic medications in youth are selective serotonin reuptake inhibitors (SSRIs) (Bushnell et al., 2018), serotonin-norepinephrine reuptake inhibitors (SNRIs), and atypical antidepressants such as bupropion and mirtazapine. These medications are frequently prescribed for children and adolescents with obsessive-compulsive disorder (OCD), anxiety disorders, and depressive disorders. In contrast, older classes (e.g., tricyclic antidepressant medications) are still commonly used for youth for neuropathic pain, migraine prophylaxis, and functional abdominal pain syndromes. Despite the widespread use of these medications, guidance on when and how to discontinue them in youth remains limited (Hathaway et al., 2017; Hosenbocus and Chahal, 2011; Strawn et al., 2023a). This is in contrast to the many studies in adults that—collectively—have examined optimal treatment durations for generalized anxiety disorder (Rickels et al., 2010) and depressive disorders, gradual and more rapid discontinuation approaches for antidepressants (Fava et al., 2015; Horowitz and Taylor, 2019; Rosenbaum et al., 1998; Tint et al., 2008), and the risk of withdrawal symptoms across medication classes (Henssler et al., 2024). Developmental differences potentially affecting the pharmacokinetics (e.g., drug-metabolizing enzyme expression) and pharmacodynamics (e.g., receptor density) of antidepressants warrant specific inquiry in the pediatric population.
To date, only a few studies have evaluated antidepressant discontinuation in youth with depressive disorders (Cheung et al., 2008; Emslie et al., 2008; Saito et al., 2023). In one study, children and adolescents 7–18 years with major depressive disorder who responded to 12 weeks of fluoxetine treatment were randomized to fluoxetine (n = 50) or placebo (n = 52) without taper for an additional 6 months. Only 42% of fluoxetine-continuing patients relapsed compared with 69% of those who were switched to placebo, and the time to relapse was significantly shorter in those randomized to placebo, with the median time to relapse of 8 weeks. In another study, escitalopram-treated adolescents with MDD were randomized to continue escitalopram or switch to placebo. During a 36-week follow-up, relapse was less likely in patients who continued escitalopram (Saito et al., 2023). Beyond these trials in youth with depressive disorders, longitudinal data, such as that from the Child Anxiety Multimodal Study Long-Term Follow-Up (Ginsburg et al., 2018; Swan et al., 2018), suggest potential benefits from 9 to 12 months of SSRI treatment for patients with generalized, separation, or social anxiety disorder. Yet, these studies generally have not addressed how to stop these medications in youth.
In deprescribing, clinicians often switch from one medication to another, either because of a lack of efficacy or side effects. This practice increasingly involves cross-titration—switching from one medication to another while overlapping the two medications. In the cross-titration of antidepressants in children and adolescents, careful consideration of specific receptor profiles and pharmacokinetics (Table 1) is paramount to mitigate the risk of relapse and discontinuation-emergent/withdrawal symptoms. Moreover, these approaches increasingly incorporate knowledge of pharmacokinetic interactions and pharmacogenetic factors that influence the metabolism of either the first or second antidepressant medication in the cross-titration.
Differentiating Withdrawal Symptoms from Relapse in Antidepressant-Treated Children and Adolescents
SSRIs, serotonin reuptake inhibitors; SNRIs, serotonin-norepinephrine reuptake inhibitors.
With these considerations in mind, we systematically reviewed studies of antidepressant discontinuation in children and adolescents as well as clinical study reports and package labeling from the Food and Drug Administration, in addition to relevant pharmacokinetic and pharmacogenetic data for SSRIs, SNRIs, and atypical antidepressants in youth. If data were unavailable, we expanded our search to include adult studies. We describe specific discontinuation-related adverse events associated with these medications based on available data, as well as pharmacokinetic and pharmacodynamically informed strategies for cross-titration of selected antidepressants.
Methods
Search strategy
Studies were obtained through an electronic search of English-language articles in PubMed (1966 through May 20, 2024), and this search was repeated on August 5, 2024. In addition, we searched the Cochrane Database and the government clinical trials registry, www.clinicaltrials.gov, using the search strategy described in Supplementary Data S1. The results were then manually limited to only include clinical trials involving children and adolescents and review articles with discontinuation data. For medications without published data in youth, the search strategy was expanded to include adult trials. Articles were excluded if there were no discontinuation data presented, acute treatment, nonantidepressant medication, neonatal/infant results, and studies in lower animals (although in vitro and lower animal studies are discussed with regard to the neurobiology of antidepressant withdrawal). The references of all eligible trials and review articles were searched for any additional studies.
Results
Our search identified 528 published articles, and 526 were screened and retrieved after excluding studies that focused on adults (n = 273) and studies that did not include any discontinuation data (n = 133), as well as the application of other exclusion criteria, 28 articles from the database search were included. In addition, 19 records were obtained through other methods (Fig. 1); of these, 14 were included. The corpus of records included 13 randomized, double-blind, placebo-controlled trials with 3026 patients. Of the 13 studies, 10 examined SSRIs, four examined SNRIs, and one examined a TCA (imipramine). Two studies involved more than one class of antidepressants. Trials ranged in duration from 4 to 35 weeks. The patients’ ages ranged from 7 to 17 years, with an average age of 12.7 ± 1.5 years. Additional details related to study identification and inclusion and exclusion are shown in the PRISMA diagram (Fig. 1).
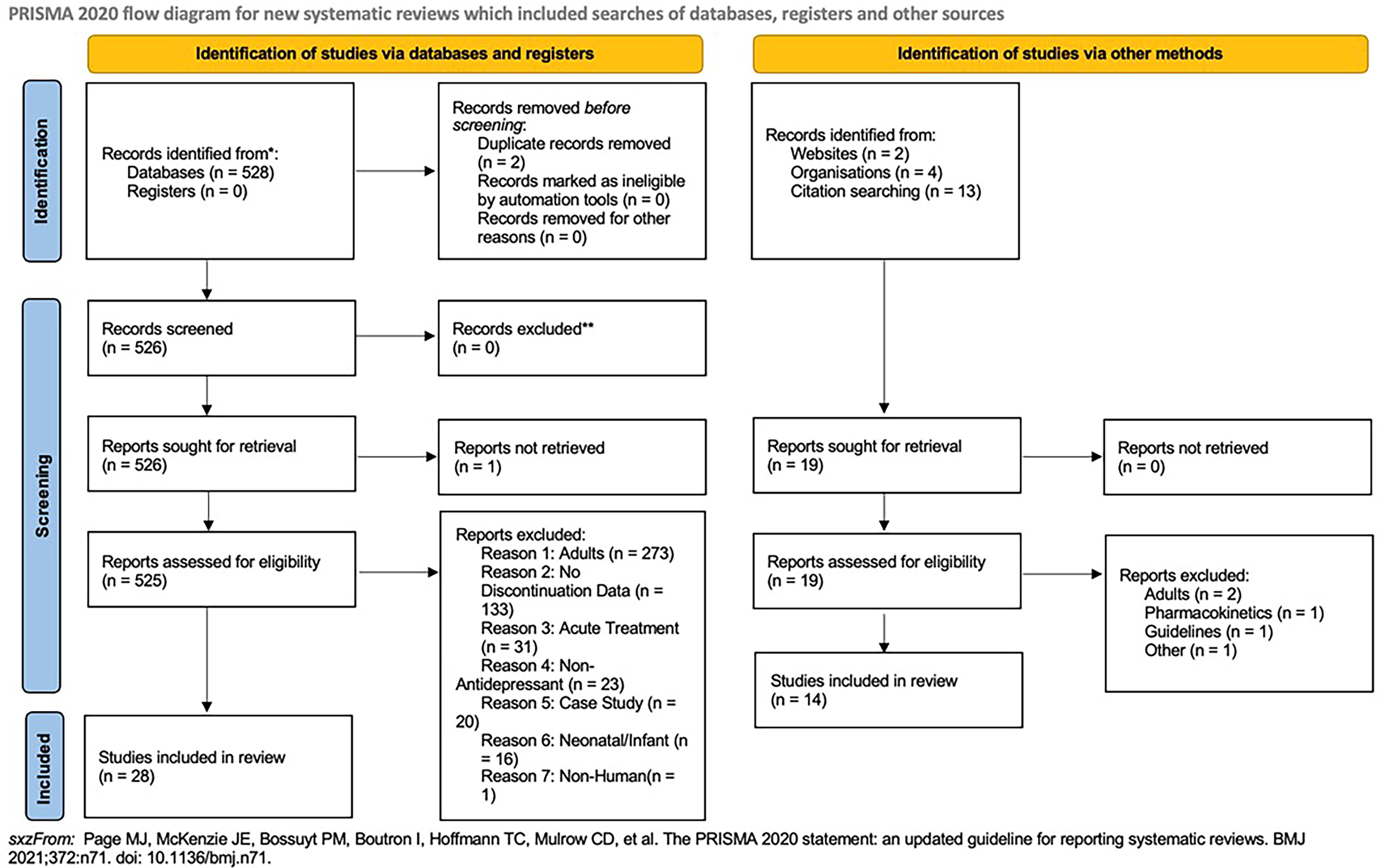
PRISMA flow diagram. This search was repeated on August 5, 2024, and no additional articles were identified.
The decision to stop antidepressant medication
The decision to discontinue antidepressant medication in a child or adolescent represents a crucial aspect of psychiatric practice, which requires a nuanced consideration of various clinical factors and flexibility in decision-making as each patient and family require a tailored approach rather than a one-size-fits-all solution. When contemplating deprescribing antidepressant therapy, clinicians must assess the child or adolescent’s level of treatment response, clinical status, comorbidities, course and duration of treatment, concurrent therapies, and family, school, and environmental factors that may have changed since treatment was initiated. These factors are important in weighing the potential benefits of continued treatment against the risks of relapse, discontinuation-emergent symptoms, and adverse effects.
Ideal candidates for antidepressant discontinuation are patients who have remitted or substantially improved, provided that they have experienced sustained relief over a period of time and that functional impairment is no longer present. Discontinuing a medication without this stability could risk undoing the progress made, and accordingly, recommendations from adults generally recognize that individuals who exhibit residual symptoms or are at high risk of relapse may benefit from ongoing antidepressant treatment to prevent symptom recurrence. Based on empirical guidance, comorbid psychiatric or medical conditions may also influence the decision to discontinue antidepressant therapy; more comorbidity may require tailored treatment approaches and potentially longer duration of treatment to effectively manage symptoms and mitigate the risk of relapse. Other factors, such as medication tolerability, side effects, and patient preferences, may also inform the decision-making process (Strawn et al., 2023a). Patients experiencing intolerable adverse effects or those who prefer nonpharmacological interventions may be candidates for tapering or discontinuation of antidepressant therapy. Conversely, individuals who have experienced minimal side effects and express a preference for continued medication may warrant ongoing treatment to maintain symptom stability and enhance quality of life.
The optimal duration of antidepressant treatment warrants careful consideration, and before embarking upon therapeutic discontinuation, children and adolescents should have had significant and sustained benefits, including remission of symptoms, for a substantial period of time. While there are limited data in the available literature regarding the optimal time course of antidepressant therapy in pediatric patients with major depressive disorder, anxiety disorders, and OCD, many clinicians continue treatment for 9–12 months after the resolution of symptoms (Hathaway et al., 2017). Emerging evidence supports that extending treatment to 12 months (Rickels et al., 2010) or longer may lower the risk of relapse illness, particularly in individuals with recurrent or chronic illness. There are no data in youth regarding differing treatment duration for those experiencing single versus recurrent mood episodes, severe symptoms, or prolonged duration of symptoms. In adults, multiple consensus guidelines suggest the need for treatment beyond 12 months and even indefinitely for depressed patients with severe symptoms (e.g., suicide attempt), episodes greater than 2 years, or multiple relapses (Keller et al., 1992; Kennedy et al., 2016).
Clinicians may also consider whether their patients are active in or have completed psychotherapy before antidepressant discontinuation. Continuation-phase or relapse-prevention cognitive behavioral therapy (RP-CBT) focuses on ameliorating remaining symptoms, promoting wellness, and preventing relapse, typically after an initial response to pharmacotherapy, and could be considered before antidepressant discontinuation. RP-CBT plus continued fluoxetine treatment was evaluated as a sequential treatment approach in treatment-responsive depressed youth (N = 144) following 6 weeks of fluoxetine (Emslie et al., 2015, 2008; Kennard et al., 2014). Over the 78-week follow-up, fluoxetine + RP-CBT reduced relapse and extended the time to relapse. Of note, clinicians should consider that psychotherapy and pharmacotherapy are not always equivalent, and caution should be exercised when contemplating replacing one with the other.
Finally, in the pediatric population, the timing of discontinuation may be particularly important with regard to school. In the Treatment of SSRI-Resistant Depression in Adolescents study, adolescents who ended their 12-week treatment during summer vacation were 1.7 times more likely to have an adequate response in depressive symptoms compared with those who ended at other times during the year (Shamseddeen et al., 2011). This suggests that there may be an added value in planning for antidepressant discontinuation during periods of reduced stress. However, further research is needed to determine if this has an impact on relapse.
Antidepressant withdrawal symptoms
While antidepressant withdrawal symptoms in pediatric patients have been described for more than four decades (Law et al., 1981), compared with adults, there is less known regarding the course of and risk factors for antidepressant withdrawal in youth (Hosenbocus and Chahal, 2011).
In adults, antidepressant dose (Fava et al., 2015; Horowitz and Taylor, 2019), duration of treatment, use for recurrent mood disorders (compared with single episodes) (Kaymaz et al., 2008), antidepressant half-life, pharmacodynamic profile (Dilsaver et al., 1983), and rate of discontinuation (Tint et al., 2008) have been associated with differences in terms of the risk of withdrawal symptoms. Across studies in youth, specific withdrawal symptoms have been reported for nearly every class of antidepressant medication, including TCAs, SSRIs, SNRIs, and atypical antidepressants. These symptoms, similar to in adults, are sometimes difficult to distinguish from relapse of the disorder being treated (Horowitz et al., 2021; Horowitz and Taylor, 2022, 2019). In general, while the magnitude of these withdrawal symptoms is greater with some antidepressants in youth (TCAs, paroxetine, and venlafaxine), there is tremendous overlap in withdrawal-associated symptoms across medications.
Withdrawal symptoms associated with antidepressants in youth vary by medication. Discontinuing SSRIs has been reported to produce palpitations, derealization, disinterest, tension, anxiety, nausea/vomiting, changes in appetite, hypersomnia, and abnormal movements. However, the magnitude of withdrawal symptoms may be related to the pharmacokinetic and pharmacodynamic profile of the medication. For example, paroxetine, known for its short half-life, muscarinic, and H1 antagonism, and potent serotonin reuptake inhibition, frequently causes more severe withdrawal symptoms, including nausea, dizziness, and abdominal pain.
Withdrawal symptoms from SNRIs in pediatric patients produce many of the same symptoms described for SSRIs. Still, they may be more characterized by dysesthesias (colloquially referred to as “brain zaps” or “electric-shock” sensations) and headaches in addition to dizziness, insomnia, abdominal pain, irritability, and nausea. However, relative to other SNRIs, venlafaxine may produce more severe withdrawal symptoms due to distinct pharmacokinetics (i.e., short half-life, Table 1). Finally, withdrawal from tricyclic antidepressants in pediatric patients can produce rebound anxiety and insomnia, primarily due to the loss of the H1 antagonism as well as cholinergic rebound, characterized by gastrointestinal distress, sweating, and urinary urgency. In addition, depressive and anxiety symptoms are frequently reported during TCA discontinuation.
Generally, withdrawal symptoms develop within hours to days of antidepressant discontinuation, although the exact timing depends on the half-life and other pharmacokinetic factors that influence metabolism. Without intervention, the natural course of antidepressant withdrawal symptoms may last for days to weeks. Child- and adolescent-specific data regarding antidepressant withdrawal courses were unable to be located at the time of this review. However, a 2022 analysis of the World Health Organization Pharmacovigilance Reporting Database, which included 31,668 reports of antidepressant withdrawal in adolescents and adults (median age 43.06 ± 14.96 years), found a median duration of 1 day with an interquartile range of 1 to 7 days, although the mean duration for “nonserious” reactions was 6 days and for “serious” was 24 days (Gastaldon et al., 2022).
Pharmacodynamics of antidepressant treatment and neurobiology of withdrawal
A comprehensive understanding of antidepressant pharmacodynamics in pediatric patients is still developing. While reward and limbic regions are relatively well developed in early adolescence, it is the development of regulatory regions in the prefrontal cortex that remains dynamic through adolescence into early adulthood and promotes executive functioning, emotional regulation, and inhibitory control. In adolescents, the SSRIs fluoxetine, sertraline, and escitalopram increase functional activity/connectivity in key regulatory regions within the prefrontal cortex (Burkhouse et al., 2018; Kujawa et al., 2016; Lu et al., 2021b, 2021a; Tao et al., 2012). In addition, changes in the connectivity of key structures within the prefrontal cortex to lower limbic structures may predict treatment response in SSRI-treated adolescents (Burkhouse et al., 2018; Kujawa et al., 2016) but not those receiving placebo (Lu et al., 2021b, 2021a). However, most data related to acute, chronic, and withdrawal symptoms associated with antidepressant medications derive from studies in lower animals or positron emission tomography (PET) studies in adults. Furthermore, there are sparse pediatric data regarding the ontogeny of pathways (e.g., receptor expression) affected by antidepressants. Thus, we lack a clear understanding of the interplay between neurodevelopment and antidepressant target engagement and molecular modulation in pediatric patients.
Long-term modulation of serotonin neurotransmission following chronic SSRI treatment and discontinuation is associated with changes in serotonin transporter and serotonin receptor expression, synaptic physiology, and neurotransmitter dynamics. SSRIs bind to and inhibit serotonin transporter (SERT) activity with varying affinities. In adults, PET studies reveal that acute and chronic SSRI administration (citalopram/escitalopram) binds in regions associated with antidepressant response (e.g., cingulate, amygdala, and raphe nuclei); however, over the initial 3 weeks of treatment, binding continues to increase in the subcortical areas (Baldinger et al., 2014). In addition, SERT occupancy in subcortical brain areas in adults correlates with plasma SSRI concentrations (Baldinger et al., 2014). Chronic SSRI exposure downregulates SERT expression and region-specific density within 3–4 weeks—a compensatory response to prolonged serotonin reuptake inhibition (Albert and Lemonde, 2004; Richardson-Jones et al., 2010; Baldinger et al., 2014). Concurrently, chronic SSRI treatment increases serotonin innervation in the prefrontal and limbic cortices (Zhou et al., 2006) in addition to inducing changes in pre- and postsynaptic serotonin (5-HT) receptor subtype expression (e.g., 5-HT1A and 5-HT2A) (Richardson-Jones et al., 2010) (Fig. 2). With chronic SSRI use, presynaptic 5-HT1A receptors become desensitized and downregulated (Fig. 2). Presynaptic 5-HT1A autoreceptor activity facilitates feedback inhibition of serotonergic neurons, modulating serotonin release and exerting regulatory control over serotonergic tone. Chronic SSRI treatment may influence 5-HT2A receptor expression and function, which may modulate synaptic plasticity and be involved in antidepressant response.

Neurobiology of serotonergic antidepressant treatment and withdrawal and the putative relationship between discontinuation rate and the emergence of withdrawal symptoms. Acute antidepressant treatment is initially associated with a reduction in the firing frequency of serotonergic neurons, which progressively increases over time. This is accompanied by a decrease in serotonin transporter (SERT) expression and 5-HT1A receptor expression. Upon discontinuation, the firing rates of serotonergic neurons rise, leading to an increase in synaptic serotonin (5-HT). During this period, 5-HT1A and postsynaptic 5-HT receptors may become supersensitive. The severity of withdrawal symptoms has been hypothesized to correlate with the extent of dose reduction (Horowitz and Taylor, 2019). Therefore, minimizing the magnitude of dose reductions may decrease the risk of withdrawal symptoms. Dotted lines represent antidepressant plasma concentrations and solid lines represent target engagement. The difference between the homeostatic set-point and antidepressant exposure (i.e., concentration over time) has been hypothesized to relate to the magnitude of risk for withdrawal symptoms. Graphs adapted from Horowitz and Taylor, 2019.
Finally, beyond serotonergic modulation, chronic SSRI exposure may also impact other monoaminergic systems, including dopamine and norepinephrine. While SSRIs primarily target serotonin reuptake inhibition, emerging evidence suggests potential cross talk between serotonergic, dopaminergic, and noradrenergic pathways, underscoring the complexity of antidepressant actions, particularly in the developing brain. Serotonergic neurons exhibit intricate connections with GABAergic-inhibiting interneurons in the reticular activating system (e.g., raphe, locus coeruleus), thus influencing noradrenergic transmission. When 5-HT2A receptors on these GABAergic interneurons are activated by the inhibition of SERT, serotonergic tone increases, suppressing norepinephrine release. Similarly, dopamine release is inhibited in the ventral tegmental area due to serotonergic influence on GABAergic interneurons expressing 5-HT2C and 5-HT3 receptors. Beyond this, α1 and D2 receptors in the reticular activating system and ventral tegmental area both enhance serotonergic activity. Antidepressants with noradrenergic activity (SNRIs) also amplify the firing of serotonergic neurons within the reticular activating system through the influence of noradrenergic innervation on serotonergic tonal activity (O’Leary et al., 2007). In contrast, acute SSRI treatment leads to a modest increase in serotonin in limbic areas as a result of the activation of presynaptic 5-HT1A and 5-HT1B autoreceptors, which counteract the effects of SERT blockade. Consequently, norepinephrine levels in the central nervous system remain unchanged, and there is a slight decrease in dopaminergic neurotransmission (Tritschler et al., 2018).
Abrupt discontinuation of antidepressants disrupts the balance of monoaminergic neurotransmission, leading to rapid withdrawal of serotonin (and norepinephrine, depending on the antidepressant) reuptake inhibition, rebound activation of the serotonergic neuron, and the subsequent alteration in synaptic 5-HT (Fig. 2) (Collins et al., 2024). This abrupt shift may boost 5-HT1A autoreceptor activity, further exacerbating the imbalance between serotonergic signaling pathways and precipitating discontinuation-emergent symptoms such as rebound anxiety, akathisia, and affective instability, in addition to the physical symptoms described below.
SSRIs
Citalopram
In a small 24-week multisite randomized placebo-controlled discontinuation study of adolescents (13–18) who responded to 12 weeks of open-label citalopram (N = 25), the time to relapse was not significantly different between groups (Cheung et al., 2016). In this trial, 75% of adolescents continuing citalopram and 62% of those who switched to placebo were without relapse during the 24-week study. Adverse effects that were significantly more common in patients who stopped citalopram included palpitations (p = 0.047) and derealization/disinterest (p = 0.019). Additional withdrawal-emergent symptoms included tension, anxiety, nausea/vomiting, and cough (Cheung et al., 2016).
Similar to escitalopram, pharmacokinetic studies of citalopram in youth suggest that its clearance is related to CYP2C19 metabolism, although this has also not been evaluated in controlled studies. Hyperbolic discontinuation (i.e., reducing dose at fixed percentages) has been proposed, although gradual discontinuation, such as that described by Cheung and colleagues (2016), represents a reasonable approach. Regarding tapering and cross-titration, when switching from citalopram to a short half-life medication, clinicians may consider initiating and titrating the new medication before discontinuing citalopram (Fig. 3).
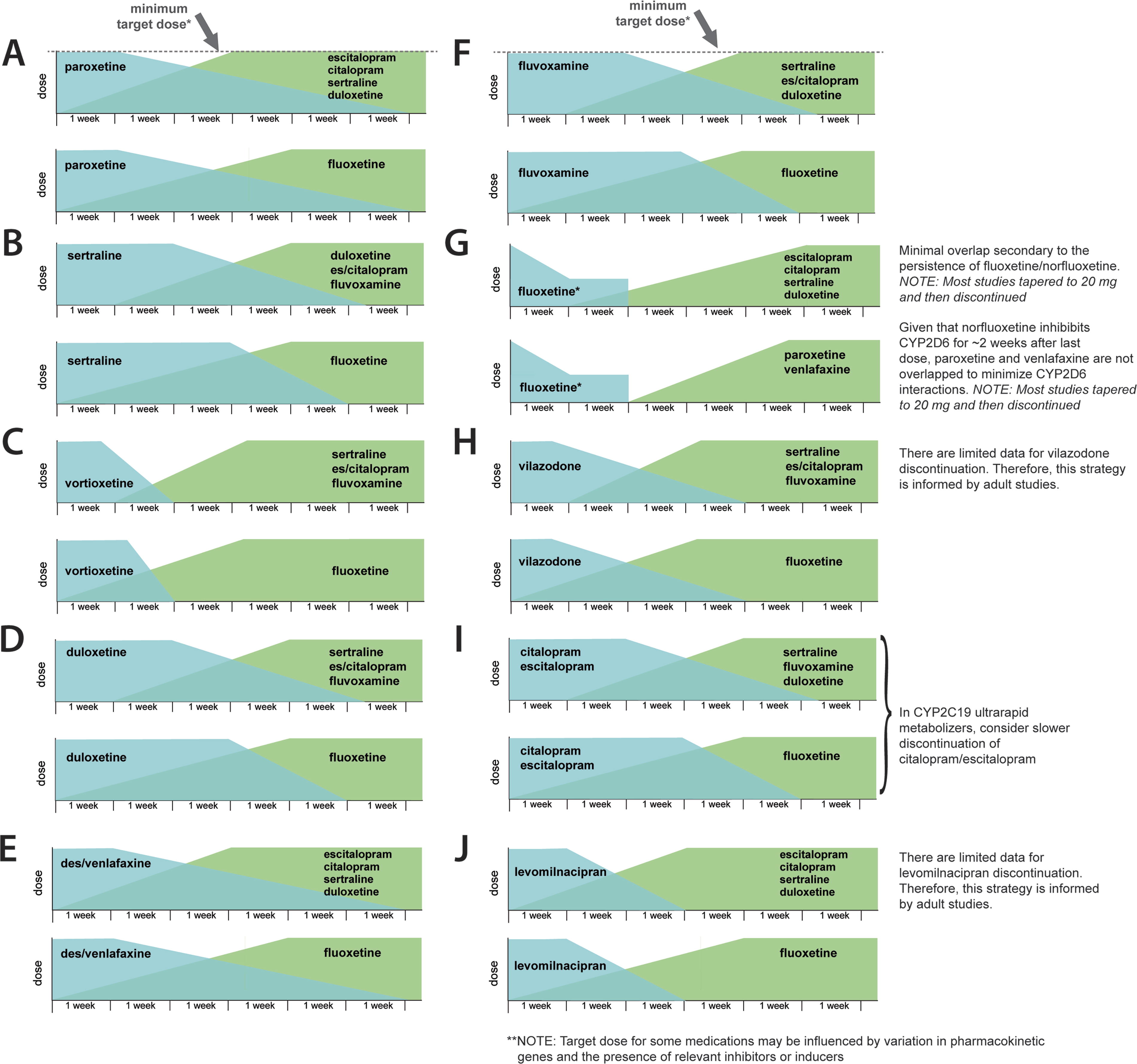
Cross-titration strategies for children and adolescents based on pharmacokinetic and pharmacodynamic properties of the first and second antidepressant. These strategies assume good tolerability of the first medication and should be reevaluated if symptoms of serotonergic toxicity or new side effects emerge during the cross-titration. In addition, clinical, developmental, and psychosocial factors should always be considered during cross-titration and deprescribing.
In the absence of prospective data, cross-titration from citalopram to another SSRI in children and adolescents without side effects should consider the time to steady state and time to achieve a “goal dose” for the second medication and the pharmacokinetics of citalopram, as described above (Fig. 3).
Escitalopram
While few studies have addressed the discontinuation of escitalopram in children and adolescents following acute treatment (Emslie et al., 2009; Saito et al., 2023; Strawn et al., 2023b, 2020), the registration trial for generalized anxiety disorder in children and adolescents included a 1-week taper, during which time youth who were treated with 20 mg daily were downtitrated to 10 mg daily and then discontinued. During this 1-week period, 8% of youth discontinuing escitalopram and 7% of those continuing placebo experienced new adverse events. One patient in each group experienced new suicidal ideation. In contrast, abdominal pain (2% vs. 0%), dizziness (4% vs. 2%), and dysesthesias (2% vs. 1%) were more common in patients discontinuing escitalopram compared with those remaining on placebo; however, these did not significantly differ between groups (Strawn et al., 2023b).
Escitalopram, as the active s-enantiomer of citalopram, corresponds to dosages that are effectively 50% of citalopram, but the pharmacokinetic parameters of the active component (s-citalopram), including half-life, are not expected to differ between escitalopram and citalopram. Pharmacokinetic modeling studies have described strategies for discontinuing escitalopram (Strawn et al., 2023a), although these studies have not considered how discontinuation strategies should be adjusted based on differences in escitalopram metabolism. Importantly, escitalopram clearance in youth is related to CYP2C19 metabolism (Strawn et al., 2019), with faster metabolizers having greater clearance and shorter half-life. Thus, slower CYP2C19 metabolizers may tolerate more rapid reductions in dose, whereas faster metabolizers could require slower discontinuation, although these approaches have not been systematically evaluated.
Studies in adults suggest discontinuation in a hyperbolic manner (Horowitz and Taylor, 2019), although this approach has not been formally evaluated in youth. In the absence of prospective data, cross-titration from escitalopram to another SSRI in children and adolescents without side effects should consider the time to steady state and time to achieve a “goal dose” for the second medication and the pharmacokinetics of escitalopram. For example, if switching to fluoxetine, initiating and titrating fluoxetine before discontinuing escitalopram would minimize the time a patient is “subtherapeutic” for both fluoxetine and escitalopram. However, a shorter cross-titration would be needed to switch to an SSRI with a shorter half-life, which requires less time to achieve a steady state (Fig. 3).
Paroxetine
In one double-blind discontinuation study of youth with major depressive disorder who were treated with paroxetine or placebo, significantly more adverse events were reported in patients discontinuing paroxetine compared with those transitioning from placebo to placebo. In this sample, discontinuation-emergent suicidality occurred in 2 of 19 patients discontinuing paroxetine compared with none of the nine patients discontinuing placebo. In addition, 15 of 19 patients experienced discontinuation-emergent worsening psychiatric symptoms compared with one of nine patients transitioning from placebo to placebo. Finally, akathisia and “withdrawal symptoms” were noted in youth discontinuing paroxetine but not in those discontinuing placebo.
A separate study of paroxetine discontinuation in children and adolescents with social anxiety disorder found that, following 16 weeks of either paroxetine or placebo, those patients who transitioned from paroxetine to placebo (compared with those transitioning from placebo to placebo) had more discontinuation-related adverse effects (Wagner et al., 2004). In this study, youth treated with >20 mg/day discontinued by tapering medication by 10 mg/day weekly over a maximum of 4 weeks. Among patients transitioning from paroxetine to placebo, 68/144 experienced discontinuation-related adverse events compared with 42/129. The most common withdrawal-emergent adverse events included nausea (16/144 vs. 3/129 for paroxetine and placebo, respectively), dizziness (16/144 vs. 2/129 for paroxetine and placebo, respectively), and abdominal pain (10/144 vs. 2/129 for paroxetine and placebo respectively) (Wagner et al., 2004).
In another study, children and adolescents treated with paroxetine (10 mg/day for 2 weeks, then 20 mg/day for 2 weeks, and then 30 mg/day for 2 weeks), dose-related increases in plasma concentrations are supraproportional in both children and adolescents (Findling et al., 2006). In this study, clearance and volume of distribution were highly dependent on paroxetine dose and CYP2D6 genotype (as well as weight) but on not age or sex and, at low doses, ostensibly before mechanism-based inhibition dominated paroxetine kinetics, there was high intraindividual variability, but this decreased as paroxetine was titrated (Findling et al., 2006).
Paroxetine—which has a markedly shorter half-life in youth than adults (Findling et al., 1999)—has remarkably complex, nonlinear pharmacokinetics and exhibits mechanism-based inhibition of CYP2D6. Paroxetine is metabolized by CYP2D6, but during this metabolism, it forms a reactive intermediate that covalently binds to the enzyme’s active site. The binding of paroxetine reactive intermediate irreversibly inactivates CYP2D6, reducing its activity (Bertelsen et al., 2003). Thus, as paroxetine is titrated, CYP2D6 activity decreases, resulting in a lower clearance at higher doses (Findling et al., 2006). However, this process accelerates as the dose is reduced. As the paroxetine dose is reduced, its clearance also increases, including in youth, which may amplify withdrawal symptoms.
Paroxetine should be discontinued slowly in youth, given its short single-dose plasma half-life of 10 hours and long plasma half-life at steady-state dosing (Table 2, Fig. 3). The rate of elimination of paroxetine from the brain in adults estimated by fluorine magnetic resonance spectroscopy (19F MRS) is equivalent to peripheral clearance, with paroxetine plus metabolite resonance dropping by 62% after a 3-day substitution of placebo from steady state, with the magnitude of brain paroxetine decrease correlating with adverse events (Henry et al., 2000). In addition, because paroxetine clearance is related to the dose (through mechanism-based inhibition), titration may be slowed as terminal doses are reached. Also, because clearance may be increased in children and adolescents at lower doses (Findling et al., 2006), it may be necessary to administer paroxetine BID or TID during the final stages of a taper. In addition, because of its anticholinergic and antihistaminergic effects, mild cholinergic rebound is possible during paroxetine withdrawal, as is some insomnia, anxiety, and agitation related to the loss of histamine-1 blockade. If cross-titrating to other CYP2D6-metabolized medications (e.g., fluoxetine or venlafaxine), the uptitration should be slow.
Pharmacokinetic Parameters of Serotonin Reuptake Inhibitors and Serotonin-Norepinephrine Reuptake Inhibitors in Pediatric Patients
Fluoxetine
The risk and timing of relapse following fluoxetine discontinuation were studied in a sample (N = 102) of treatment-responsive children and adolescents 7–18 years with major depressive disorder (mean dose = 29 mg/day), who were randomly assigned to placebo without taper versus continuation of active treatment for an additional 6 months. The relapse rate was significantly higher in the placebo group (69%; risk ratio: 2.1) versus fluoxetine (42%). The median time to relapse, as defined by significant clinical deterioration, was shorter in the placebo group (8 weeks), as was the median time to full relapse (14 weeks) compared with fluoxetine, for which time to relapse was unable to be determined as 50% of the sample did not relapse by the completion of the study (Emslie et al., 2008). Adverse events did not differ between randomized groups during the follow-up phase. In a 32-week relapse prevention phase of a double-blind, placebo-controlled, 51-week study, 20 patients continued fluoxetine at their fixed dose, and 20 were transitioned to placebo. This study switched patients directly from their fluoxetine dose (20–60 mg) to placebo without taper. Relapse occurred more quickly in patients who transitioned from fluoxetine to placebo than those who continued fluoxetine (p = 0.046) and was more common in patients who switched to placebo (34% vs. 60%). Similar to the earlier report, adverse events and tolerability were statistically similar in the F/F and F/P groups, suggesting that fluoxetine is less associated with significant discontinuation events. However, some withdrawal-emergent side effects were more common in patients switching to placebo compared with those continuing fluoxetine (headache: 15% vs. 5%, depression 10% vs. 0%; anxiety 10% vs. 0%; diarrhea 10% vs. 0%; dizziness 10% vs. 0%; myalgia 10% vs. 0%) (Emslie et al., 2004).
Although pharmacokinetic studies of fluoxetine in youth are limited, data suggest a similar profile to that of adults (Wilens et al., 2002). Of commonly prescribed antidepressants, fluoxetine has the longest half-life, ranging from 4 to 6 days, and its active metabolite, norfluoxetine, has a half-life ranging from 7 to 15 days (Wilens et al., 2002). Fluoxetine is N-demethylated to norfluoxetine by CYP2D6, and other isoenzymes, CYP2C19, CYP2C9, and CYP3A4, play a significant role in metabolism. Fluoxetine and s-norfluoxetine are potent inhibitors of CYP2D6 (competitive and noncompetitive) (Deodhar et al., 2021; Liston et al., 2002) and CYP2C19 (mechanism-based inhibition) (Deodhar et al., 2021) and moderately inhibit CYP3A4 and CYP2C9. Due to the saturation of CYP2D6, fluoxetine displays nonlinear kinetics, which accounts for increases in blood concentrations of fluoxetine that are disproportionate to dose titration. Moreover, the competitive inhibition of CYP2D6 in adults persists for 2–3 weeks following discontinuation, consistent with expected elimination (Liston et al., 2002). This is partly related to the high-affinity binding of fluoxetine and norfluoxetine to CYP2D6 and unbound fluoxetine and norfluoxetine levels circulating in the blood after discontinuation, secondary to their long elimination half-life. Using 19F MRS, brain concentrations of fluoxetine and fluorinated metabolites achieve similar levels in children as in adults, and estimates of the brain elimination half-life are similar to adults as well (16 days) (Bolo et al., 2000; Strauss et al., 2002). The slow elimination of fluoxetine from the brain and plasma contributes to the “self-tapering” of fluoxetine and has implications for the timing of cross-titration or the initiation of other psychotropic medications.
Regarding discontinuation and cross-titration, the persistent effects of fluoxetine and norfluoxetine on CYP2D6 and the long half-life generally explain the less frequent and delayed emergence of withdrawal symptoms, although not precluding their occurrence. Accordingly, in adults, headache has been reported significantly more often than placebo. At the same time, open-label studies note dizziness, lightheadedness, and dyskinesia. In general, however, discontinuation studies in adults suggest that withdrawal symptoms are mild or absent compared with other SSRIs, with abrupt discontinuation usually well-tolerated (Zajecka et al., 1998). Furthermore, in a study of adults, a 5-day interruption was not associated with discontinuation symptoms (Michelson et al., 2000). In the largest study of antidepressant discontinuation in adults, patients treated with fluoxetine 20 mg daily were discontinued by taking fluoxetine 20 mg every other day for a month and then discontinuing (Duffy et al., 2019).
In general, if no further therapy is planned, fluoxetine “self-tapers” so it can be abruptly discontinued from 20 mg/day with an observed gradual reduction in serum level over time (2–6 weeks). Pediatric studies involving patients treated with 40 mg daily have used a reduction to 20 mg and then discontinuation, and this approach appears appropriate for routine clinical practice (Fig. 3). Clinically, if changing to another medication, one approach is a washout period of 2–3 weeks followed by a recommended titration of the second antidepressant. However, a longer washout period might be appropriate if transitioning to other CYP2D6-metabolized medications, including paroxetine, venlafaxine, duloxetine, vortioxetine, and tricyclic antidepressants.
Sertraline
A 52-week multisite placebo-controlled randomized discontinuation study was conducted of treatment-responsive depressed adolescents (N = 22) aged 13–19 who completed open-label acute (12-week) and maintenance (24-week) treatment with sertraline. None of the subjects who transitioned to placebo remained without recurrence of depression, compared with 38% of those who continued sertraline. In the placebo group (N = 9), the most common adverse effects were runny nose (56%), rash/skin irritation (44%), inner tension (44%), anxiety/worry (44%), cough (33%), and less reported but still significant dry mouth (22%), nausea/vomiting (22%), diarrhea (22%), increased/decreased appetite (22%/22%), hypersomnia (11%), and abnormal movements (11%) (Cheung et al., 2008). In this study, sertraline was decreased by 25% each week for 4 weeks. Over 70% of those subjects randomized to placebo who relapsed or exited the study did so within approximately 12 weeks after substitution of placebo was begun.
In a 35-day, multiple-dosing pharmacokinetic study of sertraline in 61 children and adolescents with depression or OCD, AUC, Cmax, and elimination half-life values were reported to be similar for children versus adolescents and comparable with adult data, with t½ for sertraline of approximately 26 hours. Sertraline was titrated up to a maximum of 200 mg/day and then was openly and abruptly discontinued after 35 days. By a final pharmacokinetic sampling 9 days after discontinuation, sertraline concentrations were negligible, consistent with the empirical t½ of 26 hours (Alderman et al., 1998). No data on possible withdrawal symptoms or clinical status were included. In children and adolescents, the half-life of sertraline is dependent upon CYP2C19 activity (Poweleit et al., 2023; Strawn et al., 2019) and CYP2B6; clearance is reduced with slower CYP2C19 metabolism (Poweleit et al., 2023). Moreover, patients with increased CYP2C19 metabolism (e.g., ultrarapid metabolizers) may be more likely to experience discontinuation symptoms due to the shortened half-life of sertraline relative to other phenotypes.
Hyperbolic discontinuation has been recommended for sertraline discontinuation in youth over several months, based on studies in adults (Strawn et al., 2023a). Alternatively, the single pediatric study to evaluate discontinuation of sertraline in depressed adolescents used reductions of 25% each week for 4 weeks (Cheung et al., 2008). Notably, the largest study of antidepressant discontinuation in adults, the Antidepressants to Prevent Relapse in Depression (ANTLER) study, reduced sertraline from 100 mg to 50 mg for 1 month and then to 25 mg every other day for a month (Duffy et al., 2019). However, this approach may not be appropriate for most adults and is likely inappropriate for youth, based on the pharmacokinetics of sertraline (Poweleit et al., 2023) and the rapid relapse in depressed youth, although with a more accelerated taper (Cheung et al., 2008).
Fluvoxamine
Fluvoxamine has been systematically evaluated in pediatric patients with OCD (Riddle et al., 2001; Riddle et al., 1996) and mixed anxiety disorders (Walkup et al., 2001). However, there are limited data related to the discontinuation of fluvoxamine in pediatric patients. In adults with panic disorder, one study evaluated fluvoxamine discontinuation and found that withdrawal symptoms were noted in nearly 86% of patients following abrupt discontinuation (Black et al., 1993). In this study of 14 adults, dizziness/incoordination, headaches, nausea, and irritability were observed, and these symptoms peaked 5 days after discontinuation. In addition, one subject had a recurrence of panic, and another developed depressive and anxiety symptoms (Black et al., 1993). Using 19F MRS, brain concentrations of fluvoxamine and metabolites in adults substituted with placebo after achieving steady state demonstrated a brain elimination half-life of 56 hours, 2.4 times longer than measured plasma elimination. Half of all subjects experienced withdrawal symptoms of sweating, headaches, and nausea approximately 72–120 hours after fluvoxamine discontinuation when brain fluvoxamine concentrations had dropped to 35% of peak levels (Strauss et al., 1998). Results suggest that brain elimination rates may be more informative than plasma pharmacokinetics regarding risks of differing discontinuation schedules for fluvoxamine. In another report, investigators noted that fluvoxamine brain concentrations at steady state were not significantly different when comparing a pediatric sample with adults after adjusting for body mass (Strauss et al., 2002).
Fluvoxamine is primarily metabolized to fluvoxamine acid (Perucca et al., 1994) via CYP2D6 (Miura and Ohkubo, 2007; Spigset et al., 2001), and CYP2D6-poor metabolizers have less clearance than normal metabolizers. Additional fluvoxamine metabolites are metabolized by CYP1A2 (Carrillo et al., 1996), and fluvoxamine itself inhibits CYP1A2. One multiple-dose pharmacokinetic study of fluvoxamine in children and adolescents has shown prominent age and gender differences. Children between 6 and 11 years were noted to demonstrate higher peak plasma concentration, higher mean AUC concentration–time curves, and lower oral clearance versus adolescents (12–17 years) (Labellarte et al., 2004). Also, female children were noted to show AUC mean plasma concentrations twice that of males and higher Cmax, with clearance rates half that of male children, even after controlling for body mass. In contrast, plasma concentration–time profiles from the adolescents (compared with adults) suggest similar pharmacokinetics for the two populations. However, clearance is approximately 50% greater in adolescents than adults (even after normalizing to body weight).
In the absence of prospective trials, discontinuing fluvoxamine in pediatric patients is often guided by empirical experience. However, given its short half-life, discontinuation in the outpatient setting may best be done over several months. In addition, because its clearance is faster in youth than adults, twice daily dosing should be used, including during tapering and even at low doses.
Vilazodone
Studies of vilazodone in children and adolescents are limited and generally do not support the efficacy of vilazodone for pediatric patients with major depressive disorder (Durgam et al., 2018a; Findling et al., 2020). Moreover, there are limited data regarding discontinuation symptoms or approaches. Vilazodone is primarily metabolized by CYP3A4 with minor contributions from CYP2C19 and CYP2D6. Patients taking concurrent CYP3A4 inhibitors or inducers have altered vilazodone exposure, but the pharmacogenetic variation in all three genes does not seem to have a substantial impact (Boinpally et al., 2014; Bousman et al., 2023).
In a double-blind discontinuation study of vilazodone in adults with major depressive disorder, newly emergent side effects were similar across treatment groups (placebo, vilazodone 20 mg/day, and vilazodone 40 mg/day) (Durgam et al., 2018b). In addition, one negative pediatric efficacy study of vilazodone as a treatment for major depressive disorder in youth did not identify significant withdrawal symptoms during a 1-week taper of the medication, in which patients treated with 30 mg/day were tapered in two steps to 15 mg, then 5 mg/day and then discontinued; patients treated with 15 mg/day were tapered in one step to 5 mg/day and then stopped (Findling et al., 2020). No tolerability or discontinuation-emergent withdrawal symptoms were reported in publicly available databases or the primary article.
In general, while withdrawal symptoms were generally not noted over short discontinuation periods, when monitoring for recrudescence of depressive or anxiety symptoms, longer discontinuation periods may be appropriate for vilazodone, and discontinuation of 8–10 weeks might be appropriate and consistent with data supporting longer discontinuation periods with other antidepressant medications.
SNRIs
Venlafaxine
Discontinuation symptoms associated with venlafaxine may be more severe than those associated with SSRIs or other SNRIs. In an 8-week randomized trial of depressed adults (N = 288) treated with venlafaxine extended-release (average dose: 95 mg/day) or escitalopram (average dose 12 mg/day), discontinuation symptoms were more frequent for venlafaxine compared with escitalopram (e.g., agitation, cognitive impairment, diaphoresis, dizziness, fatigue, nausea, restlessness, tremor, and imbalance). A second study of adults with generalized anxiety disorder evaluating extended-release venlafaxine (average dose 184 mg/day), duloxetine (108 mg/day), and placebo found that over a 2-week discontinuation period, discontinuation symptoms were greater for venlafaxine compared with placebo (27% vs. 15%) but were similar for duloxetine and placebo (19% and 15% of patients).
There are few prospective trials of venlafaxine discontinuation in pediatric patients. One open-label, flexible-dose study of venlafaxine ER in children and adolescents (N = 86) with MDD examined discontinuation symptoms over a 14-day taper after 6 months of active treatment (Emslie et al., 2008). Taper/poststudy-emergent adverse events were reported by 28 subjects (33%), and the most reported events were headache (12%) and dizziness (6%). One participant reported suicidal ideation 2 days after the last dose; another was hospitalized for a suicide attempt 2 days after study discontinuation; and a third reported hostility 2 days after the last dose. Contextualization of adverse event frequency is not available due to an open-label, noncomparative study design.
Venlafaxine is metabolized by CYP2D6 to the active metabolite o-desmethyl venlafaxine, and the concentration of o-desmethyl venlafaxine depends upon the CYP2D6 phenotype. Accordingly, the Clinical Pharmacogenetics Implementation Consortium (CPIC) recommends avoiding venlafaxine in poor and intermediate CYP2D6 metabolizers, and ultrarapid metabolizers may experience more noradrenergic side effects (e.g., flushing, greater increases in blood pressure or heart rate). In addition, the half-life of venlafaxine increases with age, with a t1/2 of 8–10 hours in children 6–11 years and 11–14 hours in adolescents (Stahl and Strawn, 2024). This short half-life, relative to other antidepressants, is thought to potentiate withdrawal symptoms.
Taken together, the extant data from pediatric and adult studies suggest that discontinuation symptoms are common with venlafaxine and perhaps more severe compared with those of other antidepressants. While hyperbolic discontinuation may confer a clinically significant advantage over a linear taper for many antidepressants, this strategy is based mainly on SERT occupancy and may be inappropriate for venlafaxine. More conservative approaches may be warranted in youth, particularly given the potentially higher likelihood of treatment-emergent suicidality and shorter relative half-life. In general, discontinuation over 3 months with potentially longer tapers in some patients may be appropriate in youth, although this has not been prospectively studied.
Desvenlafaxine
An open-label, flexible-dose, extension study examining desvenlafaxine efficacy and tolerability in a sample of children and adolescents (N = 40) described discontinuation-associated adverse events. Desvenlafaxine was discontinued without taper for doses of ≤10 mg, with a 1-week taper for a dose of 25 mg daily, and with a 2-week taper for doses of ≥50 mg (Findling et al., 2014). During the taper period, one child reported headaches, and one adolescent reported depressive symptoms. In addition, in an analysis of desvenlafaxine discontinuation symptoms in adults discontinuing from fixed and flexibly dosed regimens, the most common withdrawal-related symptoms (≥5% of participants and compared with placebo) were dizziness (21.6% vs. 2.7%), irritability (9.5% vs. 4.3%), anxiety (5.6% vs. 1.6%), abnormal dreams (5.3% vs. 1.6%), fatigue (5.3% vs. 2.2%), headache (12.1% vs. 7%), nausea (14.2% vs. 4.9%), diarrhea (6.8% vs. 2.2%), and hyperhidrosis (5.3% vs. 0%). Adults taking desvenlafaxine scored significantly higher on the Discontinuation-Emergent Signs and Symptoms (DESS) checklist compared with those taking placebo (p values ≤0.028) (Montgomery et al., 2009). Notably, the discontinuation taper was relatively short (1 to 2 weeks), with adults taking 50 mg daily simply stopping the medication, while individuals taking 100 mg/day could either stop the medication or transition to 1 week of 50 mg/day and then discontinue. Patients treated with 200 mg/day decreased the desvenlafaxine dose to 100 mg for 1 week and then discontinued, and patients treated with 400 mg/day were tapered to 200 mg/day for 1 week and then 100 mg for 1 week and then discontinued the medication (Montgomery et al., 2009).
In pediatric patients, desvenlafaxine exhibits dose-related increases in CMAX and AUC, demonstrating generally linear pharmacokinetics. However, children exhibit higher clearance rates compared with adolescents. This higher clearance, coupled with a short half-life similar to that of the parent compound venlafaxine, likely contributes to a higher risk of withdrawal symptoms. Despite this, the severity of desvenlafaxine-related withdrawal symptoms in youth appears to be less than that seen with venlafaxine, possibly due to the pharmacodynamic differences between the two drugs in terms of serotonergic versus noradrenergic activity.
Desvenlafaxine should be discontinued gradually. Although pediatric trials have discontinued desvenlafaxine at a dose of 50 mg/day, some patients may require tapering to 25 mg/day. Generally, discontinuing desvenlafaxine should take place over at least 4 weeks, and based on the withdrawal-emergent symptoms systematically evaluated in adults, patients treated with higher doses (≥200 mg/day) may require slower discontinuation (Fig. 3).
Duloxetine
In the registration trial for duloxetine in children and adolescents with GAD, a 2-week taper was included, during which patients treated with duloxetine 120 or 90 mg/day were transitioned to 60 mg/day for 1 week and then 30 mg/day for 1 week. Patients treated with 60 mg/day were transitioned to 30 mg/day for 1 week. During this period, one patient experienced suicidal ideation after discontinuing duloxetine and taking a placebo. However, during this taper, no patients treated with duloxetine or placebo reported suicidal behavior or nonsuicidal self-injurious behavior. Overall, suicidal ideation was reported in two duloxetine-treated patients and one placebo-treated patient. Other taper-emergent adverse events were headache (n = 2) and upper abdominal pain (n = 2) (Strawn et al., 2015). In adolescents with depression undergoing a 2-week taper from duloxetine (30–120 mg daily), discontinuation-related adverse events that were reported by more than one patient were headache, dizziness, insomnia/initial insomnia, paresthesia, upper abdominal pain, irritability, nausea, depression, and seasonal allergies. In contrast, only headaches were reported in more than one patient who discontinued fluoxetine in this trial (Emslie et al., 2015).
This deprescribing strategy was also used in a Phase 3 multicenter trial of children and adolescents with major depressive disorder (Emslie et al., 2014; March et al., 2009; Prakash et al., 2012). Finally, in the Japanese open-label study of adolescents with major depressive disorder, patients treated with 60 mg daily were transitioned to 40 mg daily for 1 week and then 20 mg daily for 1 week (Phase-3 clinical study of duloxetine hydrochloride in children and adolescent patients with depressive disorder: An open-label extension study, https://cdn.clinicaltrials.gov/large-docs/53/NCT03395353/Prot_000.pdf). A study of adolescents with major depressive disorder-transitioned patients who were taking duloxetine 120 mg daily to 60 mg daily for 1 week and then to 30 mg daily.
Duloxetine has a relatively short half-life (∼10 hours), and clearance is generally faster in pediatric patients than in adults. Steady-state concentrations are, on average, 30% lower in children and adolescents than in adults (Lobo et al., 2014). Moreover, duloxetine metabolism may be influenced by variations in CYP2D6 and CYP1A2, although pediatric studies have been underpowered to detect the impact of CYP2D6 genetic variation on duloxetine exposure.
Based on the discontinuation strategies used in the registration trials and the available pharmacokinetic data, we recommend slower discontinuation in children and adolescents. Specifically, we recommend 2–4-week intervals between dose reductions from 90 to 120 mg. In addition, cross-titration strategies should consider the clearance of duloxetine and the absorption and time to steady state of the second antidepressant, as described in Fig. 3.
Levomilnacipran
In two double-blind studies, children aged 7–17 with major depressive disorder were treated with placebo, fluoxetine (20 mg/d), or levomilnacipran (40–80 mg/day) for 8 weeks. In the taper period, all patients on levomilnacipran reduced their dose to 40 mg/day on days 1 and 2 and then 20 mg/day from day 3 through day 7 (Radecki et al., 2024). Withdrawal symptoms were not reported, and we could not locate the clinical study report on the Food and Drug Administration (FDA) archive. In adults, the incidence of adverse events during the double-blind down-taper period was approximately 10% in both treatment patients stopping the placebo and those discontinuing levomilnacipran (Sambunaris et al., 2014).
Pharmacokinetic data related to levomilnacipran have not been published, although a review of the FDA Multi-Disciplinary Review and Evaluation, part of the new drug application, for levomilnacipran revealed that the manufacturer updated adult population pharmacokinetic models “with the sparse [pharmacokinetic] data collected in the pediatric studies… the PPK analyses have not been extensively reviewed. Based on the Sponsor’s PPK simulations, the exposure levomilnacipran in children 7 to <12 years old and adolescents (12 to <18 years) were relatively similar but slightly lower compared to adults” (NDA 204168/S-10, accessed from: www.fda.gov/media/167755/download). The pharmacodynamics of levomilnacipran are unique among SNRIs in that it has a 15-fold greater selectivity for norepinephrine compared with serotonin reuptake inhibition (relative to duloxetine, desvenlafaxine, or venlafaxine) (Auclair et al., 2013). However, at high doses, serotonergic reuptake inhibition may occur. In addition, levomilnacipran lacks affinity for other receptors, including the dopaminergic, adrenergic, histaminic, and muscarinic receptors (Auclair et al., 2013), which suggests that withdrawal-related symptoms are primarily driven by a noradrenergic and, to a lesser extent, serotonergic mechanism.
Pediatric discontinuation data are lacking; however, based on the pharmacokinetics and approach used in registration trials, shorter discontinuation may be appropriate for levomilnacipran (e.g., discontinuation over 2–4 weeks).
Atypical antidepressants
Bupropion
Published data regarding bupropion discontinuation in children and adolescents were not available at the time of this review. In adults, discontinuation symptoms are uncommon (Koshino et al., 2013; Remick et al., 1982), and a report of the European Monitoring Agency’s drug monitoring database identified only 299 of 20,720 (1.44%) adverse drug reports associated with bupropion were related to withdrawal (Schifano and Chiappini, 2018). Nonetheless, two adult case reports describe discontinuation-emergent irritability, anxiety, headache, and myalgia for one patient and acute dystonia for another (Berigan, 2002; Wang et al., 2007). For both patients, symptoms resolved upon restarting bupropion.
Bupropion is primarily metabolized by CYP2B6 (Pearce et al., 2016). Bupropion and its three major metabolites are CYP2D6 inhibitors, although at present, there are no dosing alterations based on pharmacogenetic variation recommended. Bupropion displays linear pharmacokinetics in pediatric and adult patients (Daviss et al., 2005). A pharmacokinetic study of bupropion in depressed pediatric patients (N = 16) demonstrated that responders had a higher 24-hour mean area under plasma concentration time curves (AUC0-24) for bupropion and its metabolites than nonresponders (Burleson Daviss et al., 2006). In another pharmacokinetic study of adolescents and adults, younger adolescents (12–14 years) had higher bupropion peak concentrations and AUC0-t compared with older adolescents (15–17 years) and adults, resulting in a three- to fourfold increase in total exposure in younger adolescents, reasons for which were hypothesized to be multifactorial (Oh and Crean, 2015). These pharmacokinetic observations may be considered when predicting withdrawal symptoms and risk of relapse, although the exact clinical significance is uncertain.
In addition to pharmacokinetics, bupropion pharmacodynamics are relatively favorable from a withdrawal standpoint. Bupropion inhibits the reuptake of dopamine and norepinephrine, thus it may be discontinued more rapidly, over 1–2 weeks, compared with antidepressants with prominent serotonergic effects. However, tapering more slowly may still be helpful in terms of allowing for the detection of reemerging symptoms. As bupropion is a CYP2D6 inhibitor, cross-titration to a CYP2D6-metabolized medication (e.g., fluoxetine, paroxetine, venlafaxine) should be done cautiously.
Mirtazapine
One case report detailed a 15-year-old patient who experienced hypotension requiring vasopressor support 8 hours after mirtazapine and paroxetine were held for cardiac surgery. Upon restarting paroxetine and mirtazapine, hypotension resolved, and vasopressors were discontinued (Novak et al., 2008); however, in this report, the hypotension may have been related to other postoperative sequelae rather than discontinuation of antidepressants. Other case reports in adults report induction of hypomania as well as panic attacks upon mirtazapine discontinuation, the mechanism of which was suggested to be a resulting “noradrenergic hyperactivity” (Fauchère, 2004; Klesmer et al., 2000; MacCall and Callender, 1999).
In the ANTLER study, adults taking mirtazapine 30 mg took 15 mg daily for 1 month and then 15 mg every other day (Lewis et al., 2021). Although the study did not report discontinuation-emergent adverse effects for individual antidepressants, every-other-day dosing may increase the likelihood of withdrawal symptoms, as the mean half-life of mirtazapine is 22 hours (Davis and Wilde, 1996). Every-other-day dosing may lead to adherence challenges in children and adolescents; thus, it may be reasonable to consider dose reduction from 15 mg to 7.5 mg daily rather than 15 mg every other day. This approach may minimize the incidence of withdrawal symptoms, including rebound increases in appetite and insomnia, as well as anxiety, and allows for the reemerging symptoms of the disorder being treated to be recognized and managed. In addition, while data are limited, some patients may experience rebound insomnia as a result of discontinuation of the H1 blockade associated with mirtazapine. Therefore, centrally acting H1 antagonists could be considered when discontinuing mirtazapine should rebound insomnia or anxiety emerge (Table 3).
Pharmacologic Management of Withdrawal Symptoms
Recommend avoiding trazodone if ongoing CYP2D6 inhibition is suspected, due to decreased metabolism of major metabolite meta-chlorophenylpiperazine and increased risk for side effects.
Vortioxetine
Vortioxetine discontinuation symptoms in pediatric patients have not been systematically evaluated. However, in adults, discontinuation symptoms are uncommon, unrelated to the discontinuation approach (abrupt vs. gradual), and similar to placebo (Baldwin et al., 2016; Boulenger et al., 2014; Siwek et al., 2021). A retrospective chart review of 263 vortioxetine-treated adult discontinuation symptoms occurred in 3% of patients. It included mood lability, irritability, nervousness, and agitation (Siwek et al., 2021) and were unrelated to dose or discontinuation duration (abrupt vs. gradual). In other studies, discontinuation symptoms, as measured by the DESS score, were comparable with placebo 2 weeks after abrupt discontinuation (Boulenger et al., 2014). The lack of significant withdrawal symptoms may be related to the longer half-life (66 hours) of vortioxetine and suggests that taper may not be necessary.
A multidose pharmacokinetic profile of vortioxetine in children and adolescents (N = 48) demonstrated reduced clearance and longer t1/2 (60 hours vs. 47 hours) in children compared with adolescents (Findling et al., 2017). This may be explained, in part, by significant positive associations observed between weight and volume of distribution (p < 0.01) and age and oral clearance (p < 0.01). Vortioxetine is metabolized by several CYP450 and cytosolic enzymes to inactive metabolites. In the Findling study, mean vortioxetine clearance was lower in CYP2D6-poor metabolizers (n = 2), but clinical significance and impact on tolerability and discontinuation are unknown.
In the absence of data, vortioxetine may be decreased over 4 weeks, and there appears to be a low risk of withdrawal symptoms. In addition, it may be more rapidly discontinued when a clinician wishes to cross-titrate (Fig. 3).
Tricyclic antidepressants
Several studies have evaluated the discontinuation of TCAs in children and adolescents, although these were generally—with one exception—conducted decades ago. Serotonergic and noradrenergic withdrawal symptoms are common in addition to cholinergic rebound, given many TCAs have significant muscarinic receptor binding. In general, in pediatric reports, these can be addressed by reintroducing the TCA instead of using a separate cholinergic antagonist in youth.
Amitriptyline
The data on discontinuation of amitriptyline in pediatric patients largely derive from case reports. In one case, amitriptyline was tapered following 6 months of treatment with a dosage of 150 mg/d. Less than 120 hours after discontinuation, excessive dreaming and unrestful sleep symptoms were reported (Dilsaver et al., 1983). In another case report, abrupt discontinuation (following 4 weeks of treatment at 250 mg/d) was associated with discontinuation symptoms that persisted for 60 hours and included panic attacks and “cholinergic excess.” Importantly, reinitiation of amitriptyline at 100 mg/day ameliorated these symptoms (Gawin and Markoff, 1981). Finally, two cases of nausea and vomiting were reported following abrupt discontinuation. One case was treated with 0.8 mg of atropine (Dilsaver et al., 1983), while the other received supportive therapy (Gualtieri and Staye, 1979).
Amitriptyline is metabolized via CYP2C19 to nortriptyline, and CYP2D6 hydroxylates both nortriptyline and amitriptyline. As such, pharmacokinetic variation in these enzymes should be considered as amitriptyline is deprescribed and if cross-titrating to a medication that is metabolized by or inhibits these enzymes. In addition, the CPIC recommends that in CYP2C19-poor metabolizers, a 50% reduction of the recommended starting dose and use of therapeutic drug monitoring (TDM) to guide dose adjustments in amitriptyline-treated patients who are CYP2C19- or CYP2D6-poor metabolizers. TDM may also be relevant during deprescribing as these individuals will have markedly slower clearance of amitriptyline (Hicks et al., 2017), and monitoring may allow for faster discontinuation and ostensibly lessen the likelihood of discontinuation-emergent side effects.
While rarely used for depressive or anxiety disorders in contemporary pediatric practice, amitriptyline is commonly used for migraine prophylaxis. It is generally used at lower doses (≤50 mg/day) than typically used for the treatment of depression. At these low doses, tapering may be more abrupt, potentially over 7–10 days. However, when used for affective or anxiety disorders, at higher doses, a gradual taper may minimize the incidence of withdrawal symptoms and allow for the detection of reemerging symptoms. In addition, during this discontinuation period, cholinergic rebound and anxiety/insomnia related to loss of H1 antagonism may be observed. These symptoms may be addressed by either resuming the previously prescribed dose and decreasing the dose more gradually or using centrally acting H1 antagonists or peripherally acting anticholinergics (Table 3).
Imipramine
Withdrawal symptoms associated with imipramine may be more complex than for other antidepressants, including SSRIs and SNRIs. Imipramine discontinuation has been evaluated in several pediatric reports. One study systematically evaluated imipramine discontinuation in depressed adolescents who were treated for 8 weeks with imipramine (200–300 mg, n = 32), paroxetine (20–40 mg), or placebo (n = 9) (Le Noury et al., 2015). Blinded discontinuation of imipramine was associated with nine reports of cardiovascular symptoms (e.g., arrhythmia, bradycardia, chest pain, dizziness, and tachycardia) compared with no reports of cardiovascular adverse effects in the nine patients discontinuing placebo. Similarly, more gastrointestinal symptoms, including constipation, dry mouth, diarrhea, abdominal pain, nausea, and vomiting, were observed in patients discontinuing imipramine compared with those stopping placebo. In terms of psychiatric adverse effects associated with the discontinuation of paroxetine, one patient reported suicidal ideation, and one patient reported akathisia when discontinuing imipramine; however, the psychiatric adverse events that emerged upon discontinuation of imipramine were less common than those associated with paroxetine discontinuation in the same study. Finally, headaches were more common in patients discontinuing imipramine than those discontinuing placebo (seven events in 32 patients and none in the nine patients discontinuing placebo) (Le Noury et al., 2015). In a case report, following several missed doses of imipramine at 4.8 mg/day in 15 days, one boy developed symptoms of nausea, vomiting, headache, and lethargy. In another case, following abrupt cessation of imipramine dosed at 4.4 mg/d, another boy developed symptoms of abdominal distress, nausea, loss of appetite, social withdrawal, insomnia, and became increasingly agitated, aggressive, and assaultive. In both cases, after restarting imipramine, the symptoms halted. Imipramine was then decreased over a week. Symptoms reappeared but were much less severe (Petti and Law, 1981).
In an additional study, 22 children hospitalized for depression on a high dose of imipramine (mean dose = 138 mg/d) for a mean of 41.5 days were observed for withdrawal symptoms. The tapering period was 3–10 days, with a mean of 6.4 days. Symptoms were reported by mean total symptoms per 10-day period in pretreatment, treatment, and withdrawal phases. All subjects were reported to experience at least one withdrawal-emergent adverse event. The symptoms with the most significant increase in frequency during the withdrawal phase (p < 0.001) are gastrointestinal complaints (n = 141) and drowsiness/fatigue (n = 39). Other symptoms with a significant increase in frequency included decreased appetite (n = 41, p < 0.01), tearfulness (n = 75, p < .001), apathy/withdrawal (n = 25, p < 0.03), headaches (n = 13, p < 0.03), and agitation (n = 30, p < 0.05) (Law et al., 1981).
Imipramine is converted to desipramine, which inhibits norepinephrine reuptake, and both moieties are important with regard to deprescribing imipramine. Systemic exposure to imipramine is highly variable in pediatric patients (up to sixfold). CYP2D6 and CYP2C19 are involved in clearance and pharmacogenetic variation may contribute to the variability in exposure (Hicks et al., 2017; Weller et al., 1982). Response in depressed children is exposure-related and correlated with total imipramine+desipramine plasma levels, but not with plasma levels of imipramine alone (Preskorn et al., 1982). Furthermore, while the clearance of most TCAs is generally linear, the hydroxylation pathway becomes saturated at higher plasma concentrations for imipramine (and desipramine) (Rudorfer and Potter, 1999). Thus, the clearance of imipramine (and desipramine) may be more difficult to predict when crossing the threshold between “high” and “low” doses.
Discontinuation of imipramine should be gradual, as many patients experience withdrawal symptoms. Rebound anxiety and insomnia may occur due to the loss of H1 antagonism, and gastrointestinal symptoms may emerge as a result of cholinergic rebound. Addressing these symptoms with centrally acting H1 antagonists or peripherally acting anticholinergics can be appropriate (Table 3). In addition, if cholinergic rebound leads to diarrhea, loperamide may be helpful. In the studies described above, imipramine was discontinued over 3–10 days; however, this approach may have been too rapid. Considering imipramine’s pharmacodynamic profile, the high likelihood of histaminergic and cholinergic rebound, as well as the monoaminergic-related withdrawal symptoms, a more gradual tapering over 8–10 weeks may be better tolerated. This longer discontinuation period, based on clinical experience, could help mitigate withdrawal symptoms more effectively.
Clomipramine
In one study of adults, clomipramine was abruptly discontinued following 3 months of treatment (100–300 mg/d), and withdrawal-emergent symptoms included recrudescence of depression, suicidal ideation, increased temperature, and sweating (Diamond et al., 1989). In addition, in adults with OCD (N = 18) treated for 5–27 months with clomipramine (100–300 mg), relapse of OCD was seen in 16 of 18 patients by 7 weeks after double-blind discontinuation, with increased depressive symptoms seen in 11/18 patients. Double-blind discontinuation of clomipramine occurred over a 4-day period of 50% prior dose, and then placebo only with follow up at 1, 4, and 7 weeks postswitch (Pato et al., 1988). In this study, insomnia (67%), vivid dreams (83%), and irritability (56%) were noted in the first 4 weeks after discontinuation. Depressive symptoms began to increase by week 1 with OCD symptoms beginning to clearly rise by week 4 postclomipramine discontinuation. Treatment duration before discontinuation was unrelated to the frequency or severity of withdrawal-emergent symptoms (Pato et al., 1988). In a retrospective chart review of 352 patients treated with various serotonergic antidepressants, of whom 171 underwent drug taper and discontinuation, clomipramine was associated with the highest rate of withdrawal-emergent symptoms (31%) among all other medications, with paresthesia, irritability, and insomnia as strikingly elevated versus other serotonin reuptake inhibitors (Coupland et al., 1996). There are limited data related to clomipramine-related discontinuation symptoms in children and adolescents. In one study of children, adolescents, and adults with autism (DSM-III-R criteria) aged 6–23 years, substitution with 1 week of placebo was associated with decompensation in most patients (Gordon et al., 1993), although specific withdrawal-related symptoms are unclear.
Clomipramine is primarily metabolized by CYP2C19 (Balant-Gorgia et al., 1991; Nielsen et al., 1996; De Vos et al., 2011), in addition to CYP3A4 and CYP1A2 (Nielsen et al., 1996) while its metabolite norclomipramine and clomipramine itself are also metabolized by CYP2D6. Surprisingly, despite its frequent use in pediatric patients with OCD, pharmacokinetic data for clomipramine in youth have been difficult to locate. In adults, the elimination half-life is about 20–24 hours for clomipramine and 96 hours for norclomipramine (i.e., desmethyl clomipramine, DMCMI) (Balant-Gorgia et al., 1991; Marazziti et al. 2019).
Clomipramine’s complex pharmacology includes the parent compound’s robust serotonergic reuptake inhibition, potent noradrenergic reuptake inhibition by DMCMI, and antimuscarinic, antiadrenergic, and antihistaminic effects (Millan et al., 2001; Richelson and Nelson, 1984; Tatsumi et al., 1997). A PET study of serotonin transporter (5-HTT) occupancy of clomipramine and fluvoxamine reported that a single dose of 10 mg of clomipramine occupied 80% of 5-HTT receptors, which was equivalent to a single dose of 50 mg of fluvoxamine (Suhara et al., 2003). Coupled with these multiple pharmacologic effects, the shorter half-life of clomipramine appears to contribute to a noticeably higher risk of withdrawal-emergent effects, as evidenced by case reports of severe withdrawal reactions after discontinuation of chronic doses as low as 25 mg/day (Zemishlany et al., 1992). Therefore, discontinuation of clomipramine should be approached cautiously with more gradual and extended tapering schedules, even in the context of low-dose administration, or with more rapid cross-tapering to an alternative serotonergic antidepressant, depending on the clinical need for continued treatment. Given that therapeutic drug monitoring has received some support for determining adequate clomipramine dosing for OCD and depression (Marazziti et al., 2019; Mavissakalian et al., 1990), knowledge of prediscontinuation plasma levels may aid in determining the rapidity of dose reduction and timing of tapering plans.
Discussion
Deprescribing antidepressants is an integral part of good psychiatric medication management for all clinicians, although at present, guidance on how to do so in pediatric patients is limited, and more studies are sorely needed. Based upon our literature search, controlled trials of antidepressant discontinuation in pediatric patients are sparse, and much of current knowledge is extrapolated from data in adults and inferred from pharmacokinetic studies in youth and approaches used—for some medications—in FDA-required registration trials.
The duration of antidepressant tapering requires flexibility. Indeed, there are situations in which abrupt or more rapid discontinuation is medically necessary or unavoidable, and in situations such as inpatient or intensive outpatient programs where patients are closely monitored, discontinuation can be more rapid. However, in the general outpatient management of youth with anxiety, depression, and OCD, gradual discontinuation may be the best approach to minimize withdrawal symptoms and allow clinicians to detect reemerging symptoms or recrudescence of the disorder. In addition, while evidence supporting ideal taper rates is limited, the American Academy of Child and Adolescent Psychiatry previously recommended slower discontinuation of antidepressants in their guidelines for depression in youth. In adults, the American Psychiatric Association and NICE guidelines suggest tapering therapy over at least several weeks and considering the half-life of the antidepressant (National Institute for Health and Care Excellence, 2009). This approach recognizes that antidepressants with shorter half-lives may need to be tapered more slowly. Beyond the APA and NICE guidelines, the World Federation of Societies of Biological Psychiatry guidelines recommend tapering over 4 to 6 months for long-term treated patients and further suggest that if intolerable withdrawal symptoms occur following a dose reduction, the previously prescribed dose might be reinstituted and then decreased more gradually (Bauer et al., 2015, 2013).
Generally, medications with shorter half-lives (12 hours or less) are associated with more significant discontinuation symptoms. Consequently, parameters impacting medication half-life, such as rate of clearance and CYP450 enzyme phenotype, could theoretically impact discontinuation symptoms; however, this has not been explicitly examined. For example, youth who are CYP2C19 ultrarapid metabolizers have a faster clearance of escitalopram and sertraline compared with normal metabolizers (Poweleit et al., 2023) and experience faster decreases in exposure with missed doses (Baumel et al., 2024). Slower taper may be warranted in these patients to avoid discontinuation symptoms. Alternatively, poor metabolizers may be able to titrate more quickly due to the longer half-life and slower clearance of these drugs leading to an “inherent” taper.
Hyperbolic discontinuation strategies have also been proposed (Horowitz and Taylor, 2019; Strawn et al., 2023a) based on PET data that demonstrate serotonin transporter occupancy decreases linearly as the dose is reduced hyperbolically, although this observation does not take into account pharmacokinetic factors that affect medication exposure. Authors also suggest tapering slowly and to doses “much lower than those of therapeutic minimums,” which they contend is in line with tapering regimens of other medications associated with withdrawal symptoms. Several studies have attempted to do this utilizing tapering strips that comprised daily pouches, each containing the same or lower dose of medication than the day prior (Groot and van Os, 2021). However, these approaches have primarily been tested in patients who have failed other tapering attempts and, therefore, are likely not generalizable to standard practice. Furthermore, specialized compounding pharmacies would be required to prepare such small-dose reductions precisely, which would not be practical or available to many patients.
In outlining cross-titration strategies (Fig. 3), we incorporated data from discontinuation studies in adults, pediatric and adult pharmacokinetic data, and the pharmacodynamic profiles of the individual antidepressants. In addition, we consider the time over which withdrawal symptoms have reemerged in discontinuation studies (generally a final phase in industry-funded registration trials) and when syndromic recurrence occurs in several pediatric relapse-prevention trials (Cheung et al., 2016, 2008; Emslie et al., 2008). However, these approaches should be tailored based on a patient and family’s circumstances, the pretreatment severity of patient psychopathology, the disorder being treated (depressive vs. anxiety disorders), and also potentially by variation in pharmacokinetic genes relevant to the specific medication as well as a patient’s prior experience with withdrawal symptoms that emerge when medication doses are forgotten. In fact, a history of withdrawal symptoms may guide therapeutic discontinuation approaches, given that several missed doses due to nonadherence may precipitously drop plasma concentrations in pediatric patients (Baumel et al., 2024; Strawn et al., 2021).
Furthermore, in considering cross-titration, it is important to acknowledge that not all antidepressants are created equal, especially when it comes to their efficacy in youth. This variability means that transitioning from one antidepressant to another should be approached thoughtfully, with full awareness of the potential differences in their effectiveness. It is also essential to determine when patients can be safely considered “in the clear”—that is, when they can be considered off medication without a high risk of relapse. While data are limited, the highest likelihood of relapse is generally within 3 months of antidepressant discontinuation in youth with affective disorders. Clinicians should note the potential for differences in timing to relapse based upon the properties (e.g., half-life) of the specific medication being discontinued. Lastly, choosing medications at the outset that allow for smoother transitions to other options—should cross-titration become necessary—can prevent the challenges that often arise when clinicians are forced to navigate complex cross-tapering later on. By anticipating the possibility of an inadequate response upfront, we can avoid the complications of a difficult cross-titration or significant drug–drug interactions after the fact and during cross-titration to another medication.
We primarily focused on the pharmacokinetic and pharmacodynamic factors influencing withdrawal-related symptoms, tapering, and cross-titration. However, the psychological aspects of tapering and medication discontinuation are critically important in the pediatric population and must be considered within the family context. For many patients, particularly those with anxiety disorders, cognitive factors can significantly influence their and their families’ confidence in discontinuing medication. For example, these patients often underestimate their ability to cope with distress and anxiety-provoking situations. They may have negative, even catastrophic, expectations about the likelihood and severity of withdrawal symptoms and the potential return of their anxiety disorder or related symptoms. In effect, this may lead to an expectant, “nocebo”-like response in patients who would not otherwise experience significant withdrawal symptoms, which may, in itself, necessitate a slower taper. In these cases, discussing the progress made during treatment regarding the patient’s ability to tolerate distress and anxiety, as well as improvements in functioning, can be beneficial. Reframing catastrophic expectations related to medication discontinuation using a cognitive framework can also be helpful. There may be additional psychological barriers to discontinuing antidepressant medications. Finally, addressing intolerance of uncertainty is crucial when considering the decision to stop medication, risk and timing of potential relapse, and the description of the typical time course of withdrawal symptoms. Providing anticipatory guidance on what to expect during the discontinuation process, and reassuring patients and their families about the often-transient nature of withdrawal symptoms and discussing how these symptoms will be managed collaboratively can be effective. It may be helpful for patients and their families to know that if withdrawal symptoms emerge when the medication dose is decreased, the medication can be reintroduced at a slightly lower dose. The taper can be resumed more slowly.
During cross-titration, discontinuation symptoms may be indistinguishable from treatment-emergent side effects of new antidepressants and/or recrudescence of previously remitted psychiatric symptoms (Table 1). Clinicians may examine time of onset, duration, pattern, and response to reinitiation of antidepressant to determine the etiology of symptoms. Distinct from discontinuation symptoms, few studies have described “rebound” phenomena, which one author defined as “re-emergence of symptoms of the primary disorder to a greater extent than prior to medication.” Rebound phenomena have not been well characterized, and their incidence is unknown.
There are several limitations to the current study. The most significant constraint is the lack of controlled trials on antidepressant discontinuation in children and adolescents. In addition, for some medications, pediatric pharmacokinetic data are unavailable, necessitating the use of adult data, which may not accurately represent outcomes in younger populations. Furthermore, the optimal duration of treatment for various disorders (e.g., anxiety disorders vs. OCD vs. depressive disorders) and the predictors of response and relapse remain unclear—both of which are important for planning antidepressant deprescribing. Finally, while this review examines various pharmacodynamic and pharmacokinetic factors thought to influence withdrawal, the precise clinical relevance of these factors and the extent to which they drive withdrawal symptoms remains uncertain.
Footnotes
Disclosures
Dr. Walkup receives royalties from Oxford Press and Wolters Kluwe. Dr. Robb reports AACAP honoraria, AbbVie grant support, Alkermes grant support, Eli Lilly stock in IRA, Glaxo Smith Kline stock in IRA, Johnson & Johnson stock in IRA, Lundbeck grant support and advisory board, MapLight grant support, Neuren other data safety monitoring board, NIMH grant support and other (data safety monitoring board), NCATS grant support, NICHD advisory board, Neuroscience Education Institute honoraria and travel support, Otsuka other data safety monitoring board, Pfizer stock in IRA, Syneos Health/Tetra Therapeutics Data Safety Monitoring Board. Dr. Stahl is a consultant to Acadia, Alkermes, Allergan, AbbVie, Axsome, Clearview, Done, Eisai Pharmaceuticals, Gedeon Richter, Intra-Cellular Therapies, Karuna Therapeutics, Levo Therapeutics, Lundbeck, Neurocrine Biosciences, Neurawell, Otsuka, Relmada Therapeutics, Sage Therapeutics, Sunovion, Supernus, Taliaz, Teva, Tris Pharma, and VistaGen; options in Genomind, Lipidio, Neurawell, and Delix; member of speakers bureaus for Acadia, Lundbeck, Neurocrine, Otsuka, Servier, Sunovion, and Teva. Dr. Stahl receives grant support from Acadia, Allergan/AbbVie, Avanir, Boehringer Ingelheim, Braeburn Pharmaceuticals, Daiichi Sankyo-Brazil Eisai, Eli Lilly, Harmony Biosciences, Indivior, Intra-Cellular Therapies, Ironshore, Neurocrine, Otsuka, Pear Therapeutics, Sage, Shire Sunovion, Supernus, and Torrent. Dr. McCracken has received support from GW Biosciences (Jazz Pharmaceuticals), PCORI, Think Now, and the National Institute of Health. Dr. McCracken has consulted the University of Texas System, the Coalition for Aligning Science, and Neurocrine Biosciences. Dr. McCracken has also received honoraria from the American Academy of Neurology and the American Physician Institute. Dr. Ramsey has received research support and consulting fees from BTG Specialty Pharmaceuticals, Inc. Dr. Emslie is a consultant for Xenon Pharmaceuticals Inc. Dr. Emslie receives research support from the American Foundation for Suicide Prevention, Janssen Research and Development, LLC, National Institutes of Health, Patient-Centered Research Outcomes Institute (PCORI), and the State of Texas. Dr. Strawn has received research support from AbbVie, PCORI, MindMed, and the National Institutes of Health. He has provided consultation to Alkermes, Cerevel, AbbVie, Intracellular Therapeutics, and Otsuka. Dr. Strawn receives royalties from Springer Publishing, Cambridge University Press, and UpToDate and received material support from Myriad. Dr. Strawn has also received honoraria from Medscape Live, the Neuroscience Education Institute. The other authors declare no potential conflicts of interest.
Supplementary Material
Supplementary Data S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.