
Abstract
Background:
SPTAN1 variants are thought to affect the scaffolding that protects the axonal segment of neurons as well as neuronal synapses. The SPTAN1 gene is located in the 9q34.11 genomic region and encodes the cytoskeletal protein alpha II spectrin. Epilepsy, encephalopathy, and motor neuropathy are most commonly associated with SPTAN1 variants.
Methods:
An informed consent and questionnaire were developed in order to gather information from caregivers regarding their family members’ SPTAN1 variant. Survey results are summarized descriptively, in order of frequency.
Results:
The results of a questionnaire filled out by the caregivers of loved ones who have a SPTAN1 mutation are summarized for 25 individuals, 14 males and 11 females, who have the SPTAN1 mutation.
Conclusions:
The results of this survey mirror those reported by other authors and include epilepsy, intellectual and motor delays, encephalopathy, and motor neuropathy. Additional effects of the SPTAN1 mutation reported here include absent or difficult speech, happy personality, decline in cognitive and motor skills with age, vision and hearing abnormalities, organ and skeletal effects, autoimmune diseases, and weakened immune systems.
Introduction
The nonerythrocytic α-1 (SPTAN1) gene is located in the 9q34.11 genomic region and encodes the cytoskeletal protein αII spectrin. The α-2 spectrin is the major α-spectrin expressed in non-erythroid cells and an essential protein involved in proper myelination in zebrafish (Campbell et al., 2012). SPTAN1 consists of 56 exons that encode the 2,472 amino acid αII spectrin protein. SPTAN1 has 20 repeats. The C-terminal spectrin repeats are involved in dimerization with β-spectrin (Beijer et al., 2019).
SPTAN1 variants are thought to affect the scaffolding that protects the axonal segment of neurons as well as neuronal synapses (Morsey et al., 2023; Hernandez et al., 2021). Epilepsy, encephalopathy, and motor neuropathy are most commonly associated with SPTAN1 variants (Beijer et al., 2019; Gartner et al., 2018). However, several recent articles have expanded the phenotype. Aguilera 2021 describes an individual with Angleman Syndrome with an SPTAN1 variant. Gartner et al., 2018 reports on two individuals who have altered thyroid function and encephalopathy without epilepsy and raises the possibility of spectrin-related hypoplasia and dysfunction in the cerebellum and thyroid gland, both of which are highly dependent on robust levels of normal alpha-II spectrin. Weakness, hypotonia, muscle atrophy, minor facial abnormalities, and very narrow feet are reported by Anilkumar et al., 2019, in two siblings with an SPTAN1 variant. Beijer et al., 2019 describe 10 individuals in three families who have motor neuropathy resulting from inherited heterozygous nonsense SPTAN1 variants. Morsey et al., 2023, summarize 31 individuals (age range 1 to 72 years) who have an SPTAN1 variant. Morsey et al., 2023 separated the effects of the SPTAN1 pathogenic variant into three categories: (1) epileptic encephalopathy, (2) mild developmental delay with or without epilepsy, and (3) hereditary spastic paraplegia/hereditary ataxia. This survey encompasses subjects with epileptic encephalopathy and mild developmental delay with or without epilepsy. In addition, this survey has uncovered previously unreported observations, including expressive speech deficits, visual and auditory symptoms, abnormal immune function, and regression/progression over time.
Materials and Methods
Caregiver informed consent and questionnaire
An informed consent and questionnaire were developed to gather information from caregivers regarding their family members’ SPTAN1 pathogenic variant. The informed consent and questionnaire were reviewed by an International Reviewe Board (IRB) and considered exempt. Survey participants agreed to the sharing and publishing of anonymous results. Survey questions included the presence of an SPTAN1 variant, gender, parental age, time to diagnosis, head size, epilepsy history, motor difficulties, joint swelling and pain, personality traits, autoimmune history, organ and skeletal abnormalities, and status of cognitive and motor skills (improving and declining).
Enrollment
Parents who are part of an SPTAN1 online support group (“SPTAN1” group on Facebook) responded to and filled out the questionnaire after agreeing to the informed consent.
Analysis
Survey results are summarized descriptively, in order of frequency.
Results
This is a summary of 25 children and adults affected by the SPTAN1 variants, 14 males and 11 females. There are 10 nonsense/missense (subjects 14–22, 25), 8 deletions (subjects 1–3, 8–11, 13), 4 duplications (subjects 4–7), 2 stop codons (subjects 23 and 24), and 1 insertion (subject 12) variants reported. Nine of the 15 subjects reporting epilepsy have variants close to the C-terminal. Refer to Figure 1. Two variants, p.Glu2207del (subject 11) and Asp2303_Leu2305dup (subjects 4, 5, and 6) were also reported by Morsey et al., 2023 (p.Glu2207del, patient 20; Asp2303_Leu2305dup, patient 31). There is no known duplication between the patients described in Morsey et al., 2023 and the subjects described in this survey.
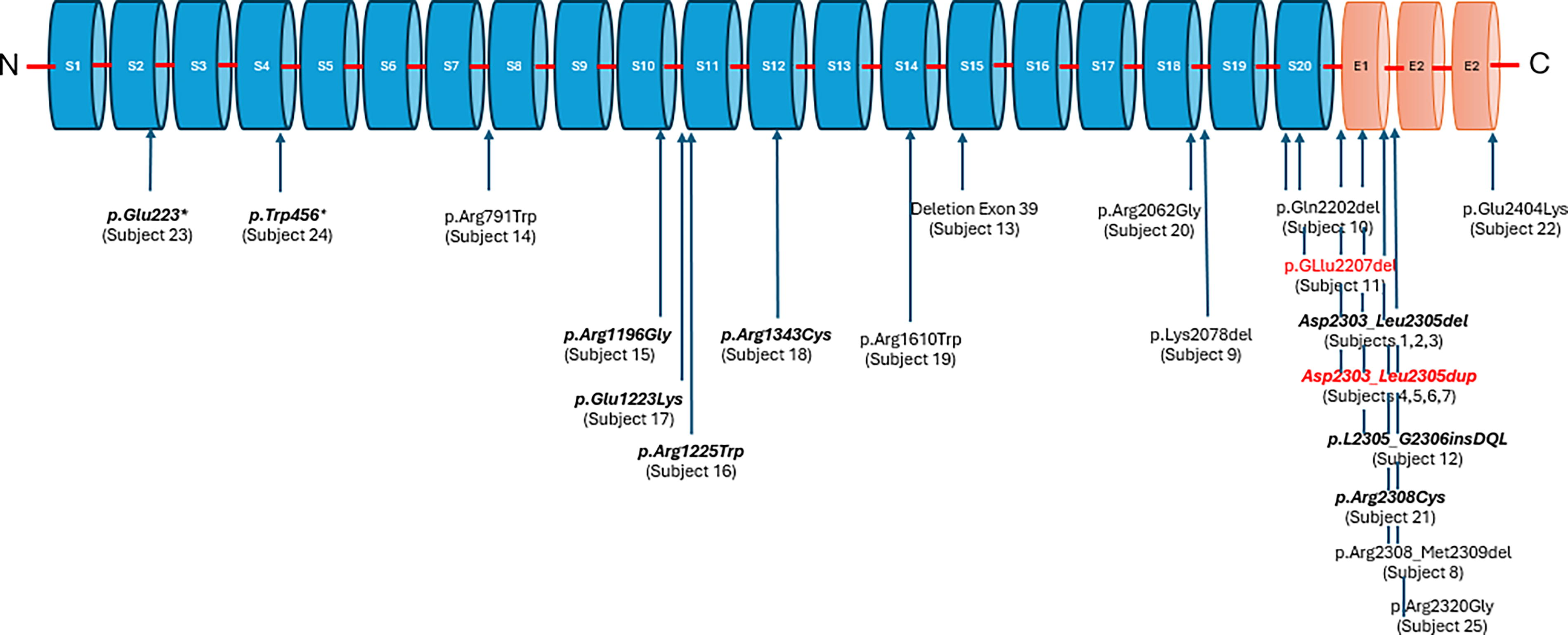
Variants reported in this survey are mapped to locations on the SPTAN1 gene. The variants in red are reported both in this survey and in Morsey et al., 2023. There is no known duplication between the patients described in Morsey et al., 2023, and the subjects who are described in this survey. Epilepsy was reported for the variants in bold italics. Subject 17’s epilepsy resolved at age 14.
The median age of the participants is 4.5 years, with a range at the time of the survey of 0.75 to 41 years. The median age when a SPTAN1 variant was diagnosed was 3.6 years, with a range of 0 to 39 years. Average gestation for the participants was 37.6 weeks. The average maternal age was 31.6 years, and the average paternal age was 33.8 years. All but three participants were born full term. Forty-two percent of mothers had a pregnancy complication, and the reported incidence of birth complications was 67%. There are no further details available regarding the types of pregnancy complications. The high rate of pregnancy complications is surprising and lends itself to further study.
De novo variants accounted for 91% of the reported SPTAN1 variants. The most common variant in this sample is c.6908_6916del (23%) and c.6908_6916dup (14%). One SPTAN1 variant was passed from father to son, and both are affected. One apparently unaffected mother has the same variant as her severely affected son (subject 16). The most commonly reported difficulties, occurring in 50% or more of the participants, included speech (absent or difficult), cognitive delays, adverse reactions to lights and noises, involuntary muscle movements, abnormal Magnetic Resonance Imaging (MRI) results, small head circumference (microcephaly), vision abnormalities, high pain tolerance, mobility difficulties, organ abnormalities, epilepsy, and GI difficulties. Refer to Table 1. Many of the same observations listed in (Table 1) have been reported by Morsey et al., 2023; Syrbe et al., 2017; Campbell et al., 2012.
Summary of SPTAN1 Survey Results as of 25 Nov 2023—N = 25 (14M, 11F)—Age Range at Time of Survey: 0.8 to 41 Years
Inherited from affected father.
Inherited from unaffected mother.
Reported in Gartner et al., 2018.
Also has an intermediate zone Fragile X variants (51 repeats).
Microcephaly as reported by caregiver. No head circumferences are available.
NA, not applicable; NR, not reported/question not answered; WC, wheelchair; UNK, unknown; A, absent; B, difficult and/or delayed; C, typical.
Reported observations not summarized in Table 1 include the following: 64% have happy and easy-going personalities. 52% reported a decline in motor and/or cognitive skills. 48% have GI difficulties (nausea, vomiting, reflux, aspiration, and constipation). 32% have bladder difficulties (frequent (UTIs and poor bladder control). 32% get sick more easily than their peers. 24% have muscle pain, hypermobile joints, or muscle cramps. 20% have joint swelling. 20% have fragile skin. 20% have slow healing. 20% have hearing changes. 20% have joint pain. 20% have blood abnormalities that include atypical blood cell morphology, intermittently high prolactin levels, anemia, and high ferritin levels. 16% have or have had an autoimmune disease (transverse myelitis; autoimmune encephalitis, vitiligo, eosinophilic esophagitis, eosinophilic gastritis, eosinophilic colitis, severe eczema, clinical euthyroidism, or multiple eosinophilic disorders). 16% have heart abnormalities (sinus ventricular tachycardia, bicuspid aortic valve, and patent foramen ovale).
Discussion and Conclusions
The parent/caregiver survey asked about all systems, and as a result, some effects not previously associated with SPTAN1 in the literature are reported here. The length of time to diagnosis ranged from 0 to 39 years with a median of 3.6 years in this cohort, most likely because genetic testing has only recently become widely available. Although SPTAN1 is most frequently associated with epilepsy and encephalopathy in the literature (Gartner et al., 2018), there are some reports that SPTAN1 is not necessarily associated with epilepsy (Morsey et al., 2023; Anilkumar et al., 2019). In this survey, just over half (60%) of the participants in this survey are reported to have epilepsy.
Beijer et al., 2019, reported that variants causing epilepsy do not seem to be linked to specific regions or domains of the SPTAN1 protein. In this study, the variants identified with epilepsy appear to be more frequent toward the C-terminus (Fig. 1) but are also found scattered throughout all the SPTAN1 repeats, in line with Beijer et al., 2019. An absence or difficulty with speech is the most frequently reported disability reported in this survey (92%). Cognitive delays were reported in 76% of the participants, and 72% have abnormal MRI findings. A high proportion (64%) of the parents/caregivers’ report that their loved one has a happy and easy-going personality.
One of the known effects of the SPTAN1 variant is microcephaly, possibly because SPTAN1 affects neurodevelopment in utero, which in turn affects head size (Wang et al., 2018; Gartner et al., 2018). In this sample, twice as many males (10/25, 40%) as females (5/25, 20%) have microcephaly (refer to Fig. 2). Hernandez et al., 2021 reported a suspected female protection factor regarding SPTAN,1 and that may be reflected in the microcephaly results here as well. In addition, of the two inherited SPTAN1 variants, one was inherited from father to son (subject 15), and both are affected. The other was inherited from mother to son (subject 16), and the son is severely affected while the mother is apparently unaffected, again raising the possibility of a female protection factor.
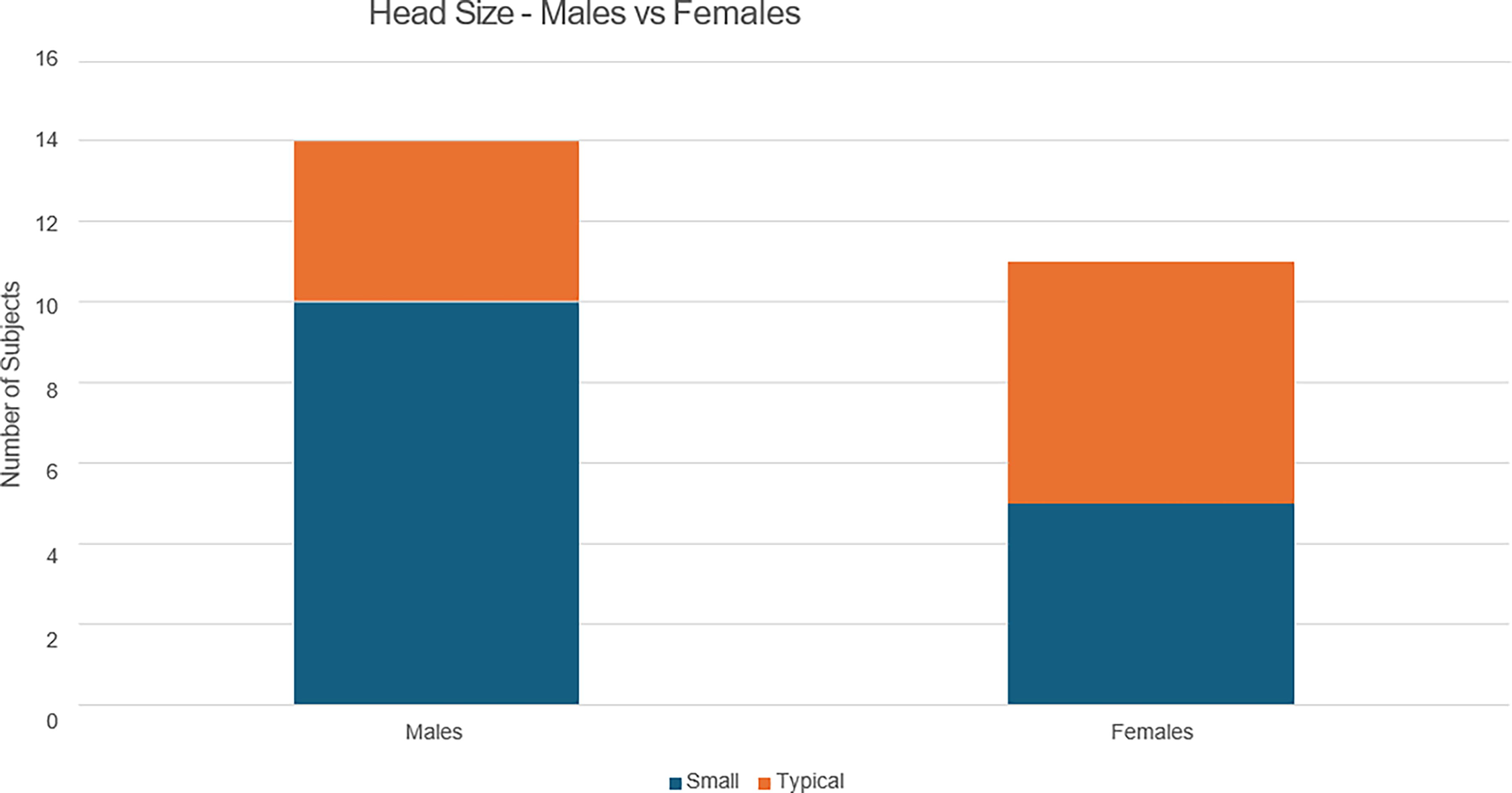
Head size—males versus females. In this sample, twice as many males (10/25, 40%) as females (5/25, 20%) have microcephaly. Hernandez et al., 2021 reported a suspected female protection factor regarding SPTAN1, and that may be reflected in the microcephaly results here as well. Actual head measurements are not available.
Just over half (52%) of the cohort are experiencing a decline in cognitive and/or motor skills with increasing age. Worsening of ataxia and involuntary muscle movements with increasing age were the most frequently reported observations in this survey, consistent with the Miazek et al., 2021 mouse model. According to Miazek et al., 2021, excessive activation of calpain proteases is a feature of neurodegenerative diseases. Miazek et al., 2021 developed a strain of C57BL/6J mice harboring a single αII spectrin point variant (Sptan1 c.3293G > A:p.R1098Q). Mice that were homozygous for this variant were embryonic lethal. Mice that were heterogeneous for this variant appeared normal at birth but shortly after developed a progressive ataxia characterized by the disruption of axonal and dendritic integrity and global neurodegeneration.
A surprising number of subjects have organ and/or skeletal abnormalities (11/25, 44%), and a third get sick more frequently than their same-aged peers. Only about 5–8% of the general population has an autoimmune disease, yet in this small cohort 16% have or have had an autoimmune disease, suggesting that SPTAN1 may affect the immune system as previously described in Meissner et al., 2017. Other difficulties reported by caregivers include bladder difficulties, muscle pain, muscle cramps, hypermobile joints, joint swelling, fragile skin, slow wound healing, hearing changes, and joint pain. Blood abnormalities, such as atypical blood cell morphology, high ferritin levels, and anemia, were also reported. Only 1 subject reported an additional genetic variant.
Subject 20 has both an SPTANI pathogenic variant and an intermediate zone Fragile X variant (51 repeats). Jalnapurka et al., 2015 and Hall et al., 2020 report that individuals with Fragile X variants in the intermediate zone (41–54 repeats) may experience some of the same effects as those with the pre- and full variant, including fragile skin, menstrual abnormalities, hypermobile joints, increased risk of autoimmune disease, tremor, and ataxia, all of which have been reported in Subject 20. Subject 20’s Fragile X intermediate zone variant and the SPTAN1 pathogenic variant may be acting synergistically or separately. It is difficult to tell how or if these 2 variants are interacting in Subject 20 given the small number of subjects in this survey and the limited data on intermediate zone Fragile X variants.
Five subjects in this survey, subjects 4–7 and 11, have pathogenic variations that are also reported in Morsey et al., 2023. Subjects 4–7 all have the p.Asp2303_Leu2305dup reported in Morsey et al., 2023, and caregivers in this survey report observations similar to those reported by Morsey et al., 2023. Subject 10 has the same p.Gln2207del pathogenic variant as individual 29 in Morsey et al., 2023, and the observations for subject 11 are also similar to those reported by Morsey et al., 2023.
Although this small cohort does not demonstrate that all the caregiver observations are associated with the SPTAN1 pathogenic variant, they are supported by other literature reports (Morsey et al., 2023; Van de Vondel et al., 2022; Morrow and Stankewich, 2021; Gartner et al., 2018), and the physician should be suspicious that SPTAN1 might be the cause of a specific observation after all other possibilities are ruled out.
The survey results are self-reported by caregivers who are part of an online support group, raising the possibility that this cohort may be more severely affected than the larger group of individuals with the SPTAN1 pathogenic variant. Parents with more severely affected children may be more likely to seek out a support group than parents of less severely affected children. Because the results are caregiver reported, the descriptions may not conform to the usual medical terms. Some of the participants reported results in a language other than English, raising the possibility of translation errors.
Footnotes
Disclosures
No competing financial interests exist.