
Abstract
Study Design:
Prospective open-label trial.
Objectives:
The objective of this study was to determine whether buspirone showed preliminary evidence of effectiveness, safety, and tolerability in individuals with Williams syndrome (WS).
Methods:
This is a 16-week, prospective, flexibly dosed, open-label trial of buspirone in 20 individuals with WS aged 5–65 years. The primary outcome measure was the Pediatric Anxiety Rating Scale (PARS).
Results:
Buspirone use (mean dose, 22.6 mg per day) was associated with a reduction in anxiety severity, with Cohen’s d estimate of −4.02 for the PARS. All 18 participants who completed the study received the Clinical Global Impression-Improvement subscale score for anxiety of “much improved” or “very much improved.” No serious or severe adverse events occurred during the trial, and no participants discontinued the study due to adverse events.
Conclusion:
Buspirone was safe and well tolerated. It was also associated with a reduction in anxiety severity. Given these findings, a double-blind, placebo-controlled study of buspirone in WS is warranted.
Introduction
Williams syndrome (WS) is a rare genetic condition that occurs in 1 out of every 7,500 live births (Strømme et al., 2002) and is caused by the deletion of 25–27 genes on chromosome 7q11.23 (Kozel et al., 2021). Common medical issues in WS include, but are not limited to, aortic stenosis, hypertension, hypercalcemia, and delayed growth (Kozel et al., 2021). The majority of individuals with WS have mild-to-moderate intellectual disability (ID) (Bellugi et al., 2000) with weaker visuospatial skills but stronger expressive language skills (Mervis and John, 2010). Individuals with WS also have characteristic facial features and are frequently described as highly sociable, friendly, and empathic, although they may also struggle to understand social norms and establish friendships (Davies et al., 1998).
Individuals with WS are also disproportionately affected by anxiety disorders, with a meta-analysis estimating the prevalence of anxiety disorders in WS to be 48% (Royston et al., 2017). Persons with WS are approximately four and 13 times more likely to be diagnosed with an anxiety disorder compared with neurotypical individuals and those with an ID, respectively (Royston et al., 2017). Generalized anxiety disorder (GAD) and specific phobias are two of the most commonly co-occurring anxiety disorders in WS (Dykens, 2003; Leyfer et al., 2006). Anxiety symptoms in WS often begin in early childhood and can persist throughout development (Einfeld et al., 2001; Leyfer et al., 2006; Woodruff-Borden et al., 2010). Studies of children, adolescents, and adults with WS have shown that untreated anxiety is associated with decreased social awareness, motivation, and isolation (Riby et al., 2014), as well as emotion recognition (Ng et al., 2016), executive functioning (Ng-Cordell et al., 2018; Woodruff-Borden et al., 2010), and adaptive functioning (Fu et al., 2015). Given the high prevalence of anxiety disorders in WS, and the negative impact that anxiety can have on social, cognitive, and adaptive functioning, it is critical to identify effective, safe, and tolerable treatments for anxiety in this population.
A handful of studies have explored the effectiveness of different behavioral interventions for the treatment of anxiety in WS, including play- and humor-infused exposure therapy (Young et al., 2023), modified group-based cognitive behavioral therapy (Thom et al., 2023), and mindfulness training (Miodrag et al., 2013). All three of these studies showed preliminary evidence of reduced anxiety severity in small samples of either children or adults with WS. Despite the importance of this work, these behavioral interventions are currently only available in highly specialized research centers and may not yet be suitable for use in the broader WS population, such as those with more severe ID and/or language impairments or psychiatric comorbidity. Therefore, there is a need to establish safe and effective treatments that are accessible to a wider range of individuals with WS.
Buspirone is an anxiolytic medication approved by the Food and Drug Administration for the treatment of GAD in adults. It acts primarily as a partial agonist on the serotonin (5-HT1A) receptor. It also has some dopaminergic activity as a D3/D4 antagonist and, to a much lesser extent, D2 antagonist (Bergman et al., 2013; Le Foll et al., 2016). Randomized controlled trials of buspirone have demonstrated reduced anxiety severity and limited adverse effects in neurotypical adults with anxiety disorders (Bohm et al., 1990). The most common adverse effects include dizziness, drowsiness, nausea, and headaches (Bristol-Myers Squibb Company, 2010).
Buspirone may be better suited for treating anxiety in individuals with WS when compared to other psychotropic medications, as it has no known cardiometabolic adverse effects, which are of particular relevance to WS (Thom et al., 2021). Previous naturalistic clinic studies and open-label trials have reported that buspirone is effective in reducing anxiety severity in other neurodevelopmental disorders, such as autism spectrum disorder (ASD) (Buitelaar et al., 1998; Ceranoglu et al., 2019), and genetic syndromes, such as Angelman syndrome (Balaj et al., 2019) and Down syndrome (Howe et al., 2022). However, only two studies have documented the use of buspirone in WS to date. Thom and colleagues (2020) examined the effectiveness of buspirone treatment in a case series of three adolescents/young adults with WS and co-occurring GAD (Thom et al., 2020). All three individuals showed a reduction in anxiety severity, as measured by the Pediatric Anxiety Rating Scale (PARS), and no adverse effects were reported. Most recently, Shin and colleagues (2024) conducted a retrospective chart review study of buspirone for the treatment of anxiety in 23 individuals with WS (Shin et al., 2024). The majority of individuals who completed a ≥16-week trial of buspirone were rated “much improved” (46%) or “very much improved” (21%) on the Clinical Global Impression-Improvement subscale (CGI-I). Adverse effects were reported in 17% of participants, but only one participant discontinued treatment due to these effects.
No prospective studies have investigated the effectiveness and safety of any psychotropic medication for the treatment of anxiety in WS. Given this, we conducted the first systematic prospective trial of buspirone for the treatment of anxiety in individuals with WS. The aims of the present study were (1) to determine whether buspirone showed preliminary evidence of effectiveness, safety, and tolerability in individuals with WS and (2) to identify which existing clinical outcome measures were the most useful for assessing response to treatment. We hypothesized that buspirone would be effective, safe, and well-tolerated in reducing anxiety severity in individuals with WS.
Methods
Participants
Eligible participants were between the ages of 5 and 65 years, had a genetic or clinical diagnosis of WS, a PARS 5-item score ≥10, and a CGI-Severity subscale (CGI-S) score of “moderately ill” or more severe (≥4). Potential participants were excluded from the study if they had obsessive-compulsive disorder, posttraumatic stress disorder, a major mood disorder, psychotic disorder, or substance use disorder; severe or profound ID based on clinical assessment and administration of a standardized IQ test; any condition that would make treatment with buspirone unsafe (e.g., allergy to buspirone, liver disease, kidney disease, and pregnancy); current use of selective serotonin reuptake inhibitors, serotonin norepinephrine reuptake inhibitors, benzodiazepines, antipsychotics, or antihistamines; a previous adequate trial of buspirone (≥20 mg per day for ≥4 weeks); or adverse reactions during a previous trial of buspirone. Concurrent use of psychotropic medications, other than those listed above, was allowed if dosing was effective, tolerable, and at an optimal dose for at least 30 days. This study was approved by the Mass General Brigham Institutional Review Board (#2021P000376). Written consent was obtained from a parent, caregiver, or legal guardian of each participant, regardless of their age. Participants with WS provided assent when able based on their cognitive and language abilities and maturity.
Clinical trial
The study was a 16-week, prospective, open-label trial of buspirone. After an in-person screen visit to determine study eligibility, participants completed a baseline visit to provide developmental, medical, and psychiatric histories; complete a standardized IQ test (Stanford-Binet Intelligence Scales-Fifth Edition, Weschler Abbreviated Scale of Intelligence-Second Edition, or Developmental Profile-Fourth Edition); and collect baseline clinical outcome measures. Follow-up visits were conducted at 4, 8, 12, and 16 weeks of treatment. Follow-up visits were completed either in-person or using a secure telehealth platform. During follow-up visits, a board-certified child, adolescent, and adult psychiatrist experienced in the care of individuals with WS evaluated participants’ therapeutic response, assessed for adverse effects, and adjusted the dosage of buspirone. Clinical outcome measures were also administered during follow-up visits. Telephone calls were conducted at 2, 6, and 10 weeks of treatment to assess for adverse effects and adjust the dosage of buspirone. The presence and severity of adverse effects were collected using a structured side effect rating scale (0 = none; 1 = mild; 2 = moderate; 3 = severe) that was completed with each participant and their parent/caregiver.
Treatment
Buspirone was prescribed to each participant’s local retail pharmacy. It was initiated at a dosage of 2.5 mg every morning, with the option to increase by 2.5 mg each week in twice-daily divided doses to a maximum dosage of 15 mg twice daily for the first 12 weeks of treatment. The decision to lower, maintain, or increase the dose was determined by the study psychiatrist based on effectiveness and tolerability. The optimal dose for each participant was reached by the end of week 12 to allow for stable dosing during the final 4 weeks of treatment.
Assessments
The following clinical outcome measures were assessed at baseline and during each follow-up visit thereafter (weeks 4, 8, 12, and 16).
Pediatric Anxiety Rating Scale
The PARS (Riddle, 2002) is a 50-item clinician-rated instrument used to assess the presence and severity of symptoms over the past week that are associated with GAD, separation anxiety disorder, and social phobia based on a child and caregiver interview. PARS 5-item severity scores, which assess the severity of endorsed anxiety symptoms, were used as the primary clinical outcome measure.
Child and Adolescent Symptom Inventory
The Child and Adolescent Symptom Inventory (CASI) is a caregiver-report questionnaire that evaluates the symptoms of various emotional and behavioral disorders (Gadow and Sprafkin, 1997). Twenty items from the anxiety subscale were administered in the present study.
Screen for childhood anxiety-related emotional disorders
The Screen for Childhood Anxiety-Related Emotional Disorders (SCARED) is a 41-item questionnaire that includes a caregiver-report version and a child self-report version (Birmaher et al., 1999). Caregivers completed the caregiver report, while the participants with WS completed the child self-report version as their cognitive ability and attention span allowed. It is used to screen for symptoms of panic disorder, GAD, separation anxiety disorder, social anxiety disorder, and school avoidance.
Aberrant Behavior Checklist, second edition—Community checklist
The Aberrant Behavior Checklist (ABC) (Aman et al., 1985) is a 58-item caregiver-report questionnaire used to assess problem behaviors in children and adults with developmental disabilities across five subscales (Irritability, Social Withdrawal, Hyperactivity, Stereotypic Behavior, and Inappropriate Speech).
Pittsburgh Sleep Quality Index
The Pittsburgh Sleep Quality Index (PSQI) is a 19-item caregiver-report questionnaire that measures sleep quality and sleep disturbances over the past month (Buysse et al., 1989).
Clinical global impression
The CGI is a clinician-rated scale commonly used in drug trials to measure illness severity and response to treatment (Guy, 1976). The CGI-S, which captures illness severity, is rated on a scale from 1 to 7 (1 = normal, not at all ill; 2 = borderline ill; 3 = mildly ill; 4 = moderately ill; 5 = markedly ill; 6 = severely ill; 7 = among the most extremely ill patients). The CGI-I score, which captures response to treatment, is also rated from 1 to 7 (1 = very much improved; 2 = much improved; 3 = minimally improved; 4 = no change; 5 = minimally worse; 6 = much worse; 7 = very much worse). The CGI-S was assessed during the screen, baseline, and last follow-up visit, while the CGI-I was assessed during each follow-up visit. The CGI was rated by the same board-certified child, adolescent, and adult psychiatrist at all visits for all of the participants. The CGI-I was designated as a prespecified secondary outcome measure. Results from the CGI-S are included for descriptive purposes.
Statistical analysis
Preliminary evidence of effectiveness required a significant decrease in mean PARS scores from baseline and a 95% confidence interval (CIs) for the effect size estimate consistent with a standardized effect size of magnitude 1.0. Preliminary evidence of safety required no observation of serious or severe adverse effects possibly, probably, or definitely associated with buspirone. Preliminary evidence of tolerability required that the 95% CIs be consistent with a dropout rate due to side effects or adverse events of 20% or less.
Analyses used an intent-to-treat approach. Sixteen-week changes in scores for all clinical outcome measures, except for the CGI-I, were estimated using repeated measures linear regression models with repeated measurements of assessment scores as outcomes and time, in categories, as the predictor. Fit was determined using restricted maximal likelihood estimation. Ninety-five percent CIs for 16-week change and tests for statistical significance of change were based on the linear contrast of the means at baseline and 16 weeks acquired from these models. Cohen’s d was calculated for each assessment by dividing the model-based estimate of 16-week change by the standard deviation (SD) of the assessment raw scores at baseline. Repeated measures linear regression models allowed unstructured covariance between repeated measurements and were fit using the PROC MIXED routine from SAS software (version 9.4).
For the CGI-I, we estimated the percentage of responders to treatment using the proportion of participants with CGI-I ratings of “much improved” or “very much improved” (CGI-I ≤2) at 16 weeks. Sixteen-week retention rate, a measure of feasibility, was estimated using the percentage of participants enrolled in the study who remained in the study for 16 weeks. Tolerability was estimated using the percentage of participants who did not withdraw from the study due to side effects or an adverse event after excluding those who discontinued participation for another reason.
Linear regression models accommodated all observed assessment scores without item-level missing data. Assessments with missing values for ≥10% of items were excluded from analysis. For the SCARED, in the presence of item-level missing data with frequency <10%, the missing values were imputed using the mean observed value for the corresponding participant and subscale. For estimating the percentage of responders to treatment, participants who discontinued treatment prior to 16 weeks due to adverse effects were classified as nonresponders, and participants who discontinued treatment for other reasons were excluded from analysis. No other missing data were imputed.
CIs for percentages, including response, retention, tolerability, and adverse effects, were calculated using Wilson’s method. CIs and statistical tests were two-sided, with test-wise alpha = 0.05. No multiple testing correction was applied.
Results
Demographics and participant characteristics
The demographic and clinical characteristics of the 20 participants with WS and their households are presented in Table 1. The mean IQ score was 66.0, with a range of 47–91. Fifty-five percent of participants were male, 10% were Hispanic, and all were White. Seventy percent were below the age of 18 years, with an age range of 6–45 years. The mean (SD) age was 17.4 (10.9) years. Fifty percent (10) received a CGI-S score of “moderately ill,” 40% (8) received a rating of “markedly ill,” and 10% (2) received a rating of “severely ill.”
Demographic and Clinical Characteristics of Participants with Williams Syndrome and Their Households
IQR, interquartile range; SD, standard deviation.
aData on approximate miles from clinic was missing for one participant.
bIQ was estimated using the abbreviated battery IQ from the Stanford-Binet Intelligence Scales, Fifth Edition (n = 17) or the cognitive scale standard score from the developmental profile 4 (n = 3).
Demographic characteristics of the participants’ caregivers are presented in Table 2. The majority of caregivers were working full-time, and 90% of maternal figures and 71% of paternal figures were college or advanced graduates.
Demographic Characteristics of Caregivers of Participants with Williams Syndrome
aOne household did not have a paternal figure living there.
bCurrent employment status was missing for one father figure. Highest occupation was missing for one mother figure and one father figure. Highest level of education was missing for two father figures.
Study flow
The flow of participants through the study is presented in Figure 1. Eighteen out of 20 participants completed the trial, resulting in an estimated 16-week retention rate of 90% (95% CI: 70%, 97%) for the study population.
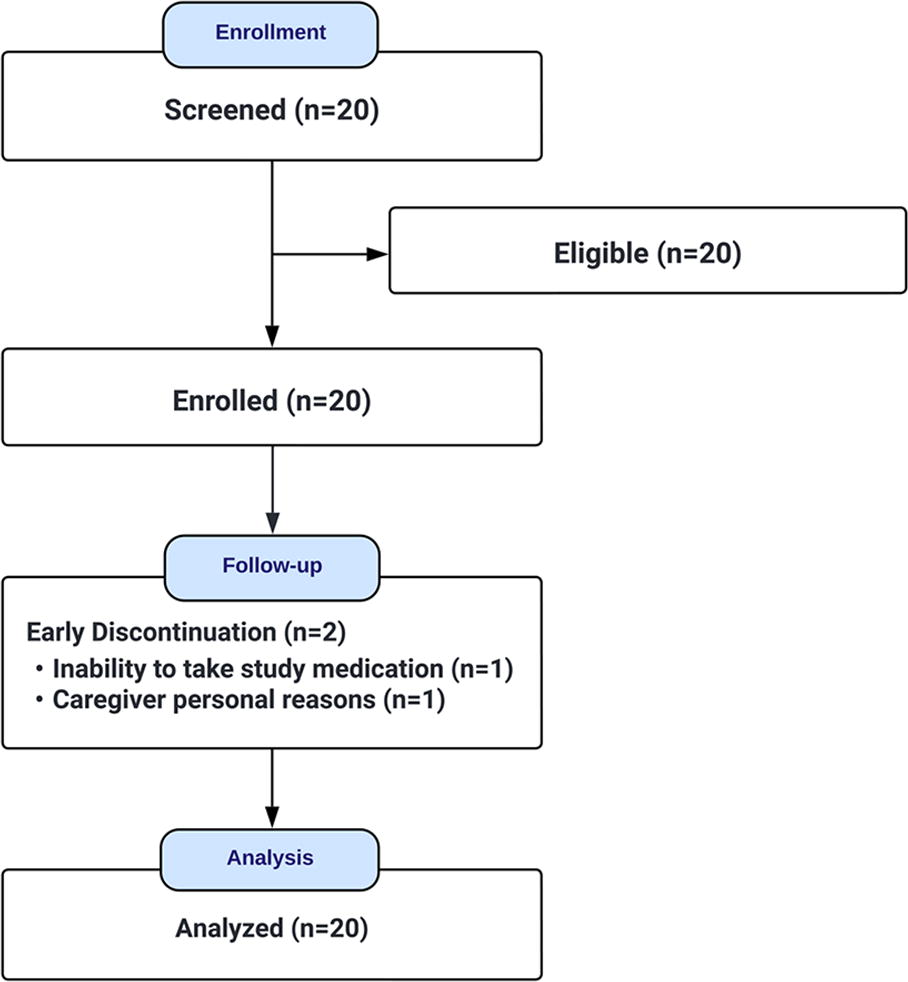
Study flow.
Dosage
Among the 18 participants who completed 16 weeks of buspirone treatment, the mean (SD) total daily dosage of buspirone was 22.6 (6.7) mg, with a median dosage of 25 mg and a range of 7.5 mg–30 mg. One participant had a recommended dosage increase at 16 weeks; the other 17 participants had no recommended dosage change at the end of the trial.
Treatment effectiveness
Changes in clinician-rated anxiety severity for each participant, as measured by the PARS, between baseline and follow-up visits are displayed in Figure 2. Sixteen-week changes in the PARS and other clinical outcome measures are presented in Table 3, along with results of corresponding statistical tests. Anxiety severity, as quantified by all three anxiety outcome measures, the PARS, CASI, and SCARED scores, decreased significantly, with Cohen’s d estimates ranging from −0.99 for the SCARED caregiver report to −4.02 for the PARS. Other behavioral symptoms, including irritability, social withdrawal, stereotypy, and hyperactivity (but not inappropriate speech), as quantified by ABC scores, decreased significantly, with Cohen’s d estimates ranging from −0.44 for stereotypy to −0.76 for irritability. Sleep disturbances, as quantified by PSQI scores, also decreased significantly, with a Cohen’s d estimate of −0.59. All 18 participants who completed the study received a CGI-I score for anxiety of “much improved” or “very much improved” (Fig. 2). Six percent (1) received a CGI-S score for anxiety of “normal, not at all ill,” 22% (4) received a score of “borderline mentally ill,” 56% (10) received a score of “mildly ill,” and 17% (3) received a score of “moderately ill.” The estimated response rate to buspirone was 100% (95% CI: 82%, 100%).
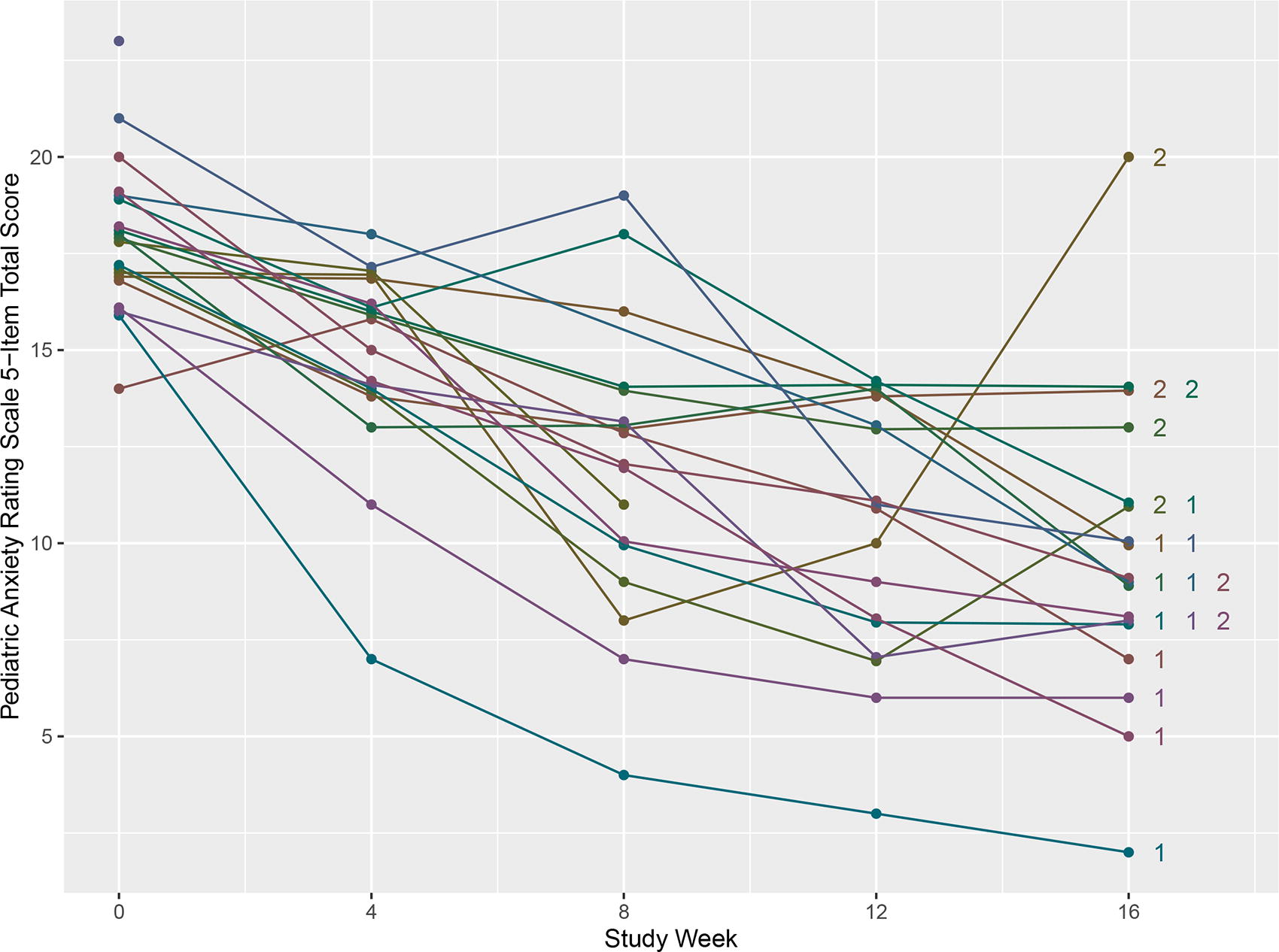
Changes in clinician-rated anxiety severity by study participant. PARS scores are plotted by week for each participant. For participants who completed 16 weeks of treatment, CGI-I scores are displayed next to plotted PARS scores at 16 weeks (1 = very much improved, 2 = much improved). Small values (≤0.2) have been added or subtracted to selected PARS scores to make it easier to differentiate treatment trajectories for each participant. CGI-I, clinical global impression-improvement subscale; PARS, Pediatric Anxiety Rating Scale.
Sixteen-Week Changes in Scores on Clinical Outcome Measures
aDue to missing data number with data analyzed is n = 9 at baseline and n = 8 at week 16 for the SCARED child/self-report; n = 17 at week 16 for the PSQI. Complete information on missing items for all clinical outcome measures is provided in Table 4.
Decreases in scores (i.e., negative values for mean 16-week change) reflect improvements in symptom severity for all assessments. Raw means and standard deviations are reported for assessment scores at baseline and week 16 of treatment. Mean 16-week change estimates, t-statistics (t), degrees of freedom (df), and p values were estimated from repeated measures linear regression models which included all non-missing assessment scores at baseline and all follow-up visits. Cohen’s d was estimated for each assessment by dividing the model-estimated mean 16-week change by the SD of the baseline assessment score.
SD, standard deviation; CI, confidence interval; PARS, Pediatric Anxiety Rating Scale (possible range: 0–25); CASI, Child and Adolescent Symptom Inventory (possible range: 0–60); SCARED, Screen for Childhood Anxiety Related Emotional Disorders (possible range: 0–82); ABC, Aberrant Behavior Checklist (possible range: Irritability: 0–45; Social Withdrawal: 0–48; Stereotypy: 0–21; Hyperactivity: 0–48; Inappropriate Speech: 0–12), PSQI, Pittsburgh Sleep Quality Index (possible range: 0–21).
Missing Data Patterns for Clinical Outcome Measures
aScores from participants with missing data for ≥10% of items were excluded from calculations of means and mean changes.
Sample sizes in the column headings reflect the number of participants enrolled in the trial at the time data were collected. nmiss,1 is the number of enrolled participants who did not complete the corresponding assessment at that specific follow-up visit. nmiss,2 is the number of enrolled participants who completed the assessment but had missing data for one or more items; the number of missing items for that corresponding assessment and participant are listed in parentheses. Only items directly contributing to the calculation of total scores for each assessment are included in this table.
PARS, Pediatric Anxiety Rating Scale; CASI, Child and Adolescent Symptom Inventory; CARED, Screen for Childhood Anxiety Related Emotional Disorders; ABC, Aberrant Behavior Checklist; PSQI, Pittsburgh Sleep Quality Index.
Clinical outcome measure performance
All clinical outcome measures that assessed anxiety severity were associated with Cohen’s d estimates of magnitude ≥0.99. The PARS was associated with the greatest Cohen’s d magnitude of 4.02 (Table 3). Information on missing data for clinical outcome measures is presented in Table 4. The PARS was completed for all enrolled participants at all time points, with the exception of missing data from one participant at the 8-week follow-up visit; this participant remained enrolled in the study for 16 weeks of treatment. No more than two enrolled participants were missing data at any time point for all clinical outcome measures, except for the SCARED child/self-report, which was completed for no more than 10 participants at any time point. The SCARED was the only clinical outcome measure to show item-level missing data.
Adverse events
No serious or severe adverse events occurred during the trial. The frequencies of all recorded adverse events and associated 95% CIs are reported in Table 5. The most commonly reported adverse events were diarrhea, appetite decrease, stomach or abdominal discomfort, and nasal congestion or cold. No participants discontinued the study due to side effects or adverse events. The estimated tolerability of a 16-week course of buspirone treatment was 100% (95% CI: 82%, 100%).
Frequency of Adverse Effects
CI, confidence interval.
Discussion
This report represents the first systematic study of any psychotropic medication for the treatment of anxiety in WS. Results of this prospective open-label study met prespecified criteria for preliminary evidence of the effectiveness, safety, and tolerability of buspirone for the treatment of anxiety in persons with WS. Cohen’s d estimates of standardized effect size were largest for the clinician-rated severity scale (PARS Cohen’s d: −4.02), intermediate for the self-report measure (SCARED Cohen’s d: −1.67), and smallest for caregiver report measures (CASI Cohen’s d: −1.03; SCARED Cohen’s d: −0.99). The PARS is a clinician-rated anxiety severity score that integrates patient and caregiver reports. It may also have been associated with the largest effect size because, unlike the SCARED and CASI scores, which represent the sum of the number of anxiety symptoms endorsed, the PARS score is based upon five severity indicators (symptom frequency, symptom severity, avoidance, interference at home, and interference outside of the home), rather than the endorsement of specific symptoms of anxiety. This is of particular relevance to WS, as patients with developmental disabilities likely have distinct clinical presentations of anxiety that are not fully captured by standard rating scales developed for and validated in the general population. Therefore, in the absence of genetic syndrome-specific measures, those focused on reducing symptom severity, rather than the elimination of specific anxiety symptoms, may be most sensitive to detecting change with treatment. Furthermore, the results support the use of the PARS for assessing improvement in anxiety symptoms in individuals with WS in future trials. Altogether, these data suggest that buspirone may be beneficial for the treatment of anxiety in WS and therefore warrant further study in a randomized controlled trial.
The findings of reduced anxiety symptom severity with buspirone treatment are consistent with previously published reports on the use of buspirone in WS, as well as in other neurodevelopmental disorders. Our group previously described a case series of three adolescents/young adults with WS who experienced a sustained response to buspirone for the treatment of GAD (Thom et al., 2020). Likewise, we recently reported on 24 patients with WS who received treatment with buspirone for anxiety under naturalistic clinic conditions (Shin et al., 2024). The response rate in this retrospective chart review study was 67% (95% CI: 47%, 82%), which is lower than the 100% response rate observed in this open-label trial (95% CI: 82%, 100%). The higher response rate observed in the present open-label trial may be due to excluding patients with other psychiatric co-morbidities, nonspecific therapeutic effects from participating in a trial (e.g., frequent clinician contact), and/or placebo response. Of note, patients with ID may experience an enhanced placebo response compared with neurotypical patients (Jensen et al., 2017). Gastrointestinal side effects were the most common type of adverse effect observed both in our retrospective chart review study and in this open-label trial. Findings from the present study are similar to those reported in an open-label trial of buspirone in psychiatrically hospitalized youth with ASD, which demonstrated a 76% response rate (Buitelaar et al., 1998).
Previous findings indicate that the response rate to buspirone may be less consistent in neurotypical populations. For example, pooled analyses of two large randomized controlled trials of buspirone in children with a primary diagnosis of GAD showed that response to buspirone did not differ from placebo (Strawn et al., 2018). Furthermore, drop out due to treatment-emergent adverse effects was higher in children treated with buspirone compared with placebo (Strawn et al., 2018). Randomized controlled trials of buspirone for the treatment of anxiety in neurotypical adults have demonstrated a response rate ranging from 38% to 67% (Feighner et al., 1982; Pecknold et al., 1989). Further research is needed to determine whether individuals with WS and other neurodevelopmental disorders or genetic syndromes have a higher response rate to buspirone than neurotypical individuals.
Our findings add to a small literature on pharmacotherapy in WS. Very few studies have investigated the effectiveness and/or safety of psychotropic medications for anxiety in WS, apart from a handful of retrospective chart reviews and case series (Green et al., 2012; Shin et al., 2024; Thom et al., 2020), despite the fact that many individuals with WS are prescribed medications to manage their anxiety. In a caregiver-report study on the prevalence of psychotropic medication use in individuals with WS, Martens and colleagues (2012) found that commonly prescribed medications included selective serotoni reuptake inhibitors (SSRIs; 24%), non-SSRI anxiolytics/antidepressants (12%), and antipsychotics (10%) (Martens et al., 2012). The majority of caregivers found these medications to be helpful or somewhat helpful in reducing their child’s anxiety symptoms (SSRIs = 81%, non-SSRI anxiolytics/antidepressants = 64%, antipsychotics = 72%). Green and colleagues (2012) examined the use of SSRIs (fluoxetine or citalopram) in a longitudinal study of five individuals with WS and a co-occurring anxiety disorder. Four out of the five individuals showed anxiety symptoms that were “much improved” with treatment based on CGI-I scores (Green et al., 2012).
This study presents data of clinical relevance, including dosing strategies, time to response, and safety. The final total daily dosage of buspirone was 22.6 mg, with a range of 7.5–30 mg. Although time to response varied, most participants showed a reduction in PARS scores between eight and 12 weeks of treatment, and all experienced a decrease in PARS scores within the first 4 weeks of the trial. No serious or severe adverse effects occurred, adding to the existing literature supporting the safety of buspirone in this medically vulnerable population. Among the secondary clinical outcome measures assessing other behavioral symptoms, we found significant reductions in four of the five subscales of the ABC (Irritability, Social Withdrawal, Stereotypy, and Hyperactivity), as well as the PSQI, during treatment. This finding suggests that buspirone may either exert nonspecific effects that improve a variety of behavioral symptoms, including anxiety, and/or that anxiety may manifest with a broad range of behavioral symptoms, including sleep disturbances, that improve with buspirone treatment.
Limitations of the current study design need to be considered when interpreting the findings of this report. As an open-label trial, this study lacked a comparator group, and scores on clinical outcome measures may be biased due to expected improvements in anxiety symptoms. Furthermore, participants were heterogeneous with respect to the presentation of their anxiety symptoms and co-occurring anxiety disorders. Other limitations include the wide age range of participants included in the study, the small sample size, and the lack of racial/ethnic and socioeconomic diversity of participants.
Despite these limitations, the present study reports findings of clinical relevance, given the large proportion of individuals with WS who experienced improvements in anxiety symptoms with the use of a safe, well-tolerated medication. Having met or exceeded our prespecified criteria for preliminary evidence of effectiveness, safety, and tolerability, the findings support the conduct of a larger, double-blind, randomized controlled trial investigating the efficacy of buspirone for the treatment of anxiety in WS.
Footnotes
Acknowledgment
M.P. is supported by the Landreth Fellowship.
Authors’ Contributions
R.P.T. contributed to the conceptualization, methodology, investigation, writing (original draft, review and editing), supervision, project administration, and funding acquisition. D.R. contributed to investigation and writing (review and editing). M.P. contributed to writing (original draft, review and editing). J.M. contributed to investigation and writing (review and editing). C.R. contributed to conceptualization, methodology, formal analysis, data curation, visualization, and writing (original draft, review and editing). C.J.M. contributed to conceptualization, methodology, resources, writing (review and editing), funding acquisition, and supervision.
Disclosures
R.P.T. receives royalties from Oxford University Press and Springer Publishing. C.J.M. is a consultant for Acadia Pharmaceuticals and receives royalties from Oxford University Press and Springer Publishing. The remaining authors have no conflicts of interest to disclose.