
Abstract
Objective:
Sedum sarmentosum Bunge, a Chinese herb, is mainly used for the treatment of chronic viral hepatitis in China, The aim of this work was to examine the cytotoxic activity of S. sarmentosum (aqueous extract, AE) against a human hepatoma cell line (HepG2) in culture.
Materials and Methods:
Cell-proliferation ability was determined by the MTT method. Cell-cycle changes and earlier period apoptotic rate of HepG2 cells were detected by flow cytometry. Apoptosis of cultured HepG2 cells induced by AE were observed with a classic laddering pattern on agarose gel electrophoresis. The mRNA levels of Bcl-2 and vascular endothelial growth factor (VEGF) were determined by reverse-transcriptase polymerase chain reaction. The protein expressions of Bcl-2, VEGF, and p-STAT3 were valued by immunocytochemistry.
Results:
The application of AE to the HepG2 cell culture caused a significant, dose-dependent inhibition of cancer cell growth. It was found that the AE treatment induced apoptosis of the cancer cells, although no changes were found after AE treatment for 48 hours in the HepG2 cell cycle. The mRNA and protein expressions of Bcl-2 and VEGF and the protein level of p-STAT3 were significantly decreased after the AE treatment for 48 hours.
Conclusions:
This study suggests that AE of S. sarmentosum has potential in preventing and inhibiting effects on hepatocellular carcinoma, which is associated with apoptosis of the cancer cells.
Introduction
S edum sarmentosum Bunge is a perennial herb widely distributed throughout China and has been traditionally used for treating various types of hepatitis. It may inhibit the production of exudates induced by inflammation, decrease the level of serum alanine aminotransferase (ALT), and show remarkable hepatoprotective activity. 1 –5 Although MeOH-soluble alkaloids isolated from this plant are considered to have antiproliferative effects on murine and human hepatoma cell lines, 6 many investigations showed that the water-soluble sarmentosin is considered the more active constituent in hepatitis therapy of this plant. 1 In addition, aqueous extraction, a traditional method of extraction, could protect the active constituent to the utmost extent. Thus, the mechanisms of hepatoprotective activities and other bioactivities of aqueous extract from this herb remain to be established.
Hepatocellular carcinoma (HCC), one of the most common malignant tumors worldwide, is the most fatal form of cancer. 7 –11 It is well known that HCC is closely related to hepatitis, especially those induced by HBV or HCV. 12 –16 In view of the therapeutic efficacy of S. sarmentosum in hepatitis and the relationship between hepatocellular carcinoma and hepatitis, we decided to extend the investigation on the antitumor activity of S. sarmentosum. Hep G2 is an established human hepatocarcinoma cell line, which was derived from the liver tissue of a 16-year-old male patient with a well-differentiated HCC. These cells are usually used for a variety of biochemical and cell-biologic studies, especially for the in vitro study of HCC. The aim of this study was to confirm the inhibiting effects of aqueous extract (AE) from S. sarmentosum on HepG2 cancer cells and investigate how AE influences the status of HepG2 cells.
Materials and Methods
Preparation of the plant extract
Aqueous extraction was conducted as the traditional method of extraction by adding 500 mL of distilled water to 50 g of dried plant of S. sarmentosum in a flask, followed by steeping in a water bath at 100°C for 30 minutes. The mixture was filtered and the residue was extracted in duplicate under the same conditions. Then, the filtrates were combined and concentrated to 100 mL. After the solution had cooled, the extract was carefully decanted away from the residual solids and centrifuged for 10 minutes at 3000 rpm. Subsequently, the supernatant was evaporated under vacuum, lyophilized to get AE powder of S. sarmentosum, and then stored at 4°C until further assay. For all subsequent experiments, AE was diluted with culture medium to determine concentrations and was sterilized by filtration. The concentration used in the experiment was based on the dry weight of the extract (μg/mL).
Cell line and culture condition
The human hepatoma HepG2 cell line was obtained from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 medium, supplemented with 10% heat-inactivated fetal bovine serum (FBS) in an incubator (Sanyo, Japan), with a humidified atmosphere of 5% CO2 in air at 37°C.
Cell proliferation assay
Cells were plated in 96-well flat-bottomed plates at 1 × 105 cells/well. After 4 hours of incubation, AE was added to the wells to get the final concentrations of 25, 50, 100, and 200 μg/mL, respectively. To the control groups was added the same volume of culture medium. Then, cells were incubated for 24, 48, and 72 hours. After that, 10 μL of 5 mg/mL of MTT (Sigma) in phosphate-buffered saline (PBS) was added to each well and incubated for an additional 4 hours at 37°C. The medium was removed and formazan was dissolved in 150 μL of dimethyl sulfoxide (DMSO), and the optical density was measured at 570 nm, using a bioassay reader (BioTek ELx800; BioTek Instruments, Winooski, VT).
Cell-cycle analysis by flow cytometry
HepG2 cells were seeded in a 24-well plate (1 × 106 cells/well) and exposed to AE at final concentrations of 50, 100, and 200 μg/mL while culture medium was added to the control group. After treatment for 48 hours, cells were harvested in PBS and fixed in ice-cold 70% ethanol overnight at 4°C and then treated with staining solution (50 μg/mL of RNase and 100 μg/mL propidium iodide [PI]) in PBS) at 4°C for 30 minutes. The stained cells were run through a FACScan (Becton Dickinson, San Jose, CA). The proportion of nuclei in each phase of the cell cycle was determined from using WinMDI DNA analysis software.
Assay of early apoptotic cells by Annexin V–FITC/PI
For apoptosis studies, HepG2 cells were incubated with AE at the indicated concentration for 48 hours. The adherent and floating cells were combined and suspended in binding buffer (10 mM of HEPES [pH 7.4], 140 mM of NaCl, and 5 mM of CaCl2). Then, cells were stained with fluorescein isothiocyanate (FITC)-labeled Annexin-V and PI, which could exclude late apoptotic and necrotic cells. After that, all stained cells were measured with FITC-PI flow cytometry (FACScaliber Becton-Dickinson) to differentiate apoptotic (Annexin V–positive and PI-negative) from necrotic cells (Annexin V– and PI-positive). Electronic compensation of the instrument was done to exclude overlapping of the emission spectra. A total of 10,000 events were acquired, and the cells were properly gated for analysis.
Analysis of DNA fragmentation
DNA fragmentation was assayed by agarose gel electrophoresis (AGE), as previously described. 17 Briefly, following 48 hours of treatment by AE, 1 × 106 cells were harvested by centrifugation, washed twice with PBS, and then lysed in 0.25 mL of ice-cold lysis buffer for 15 minutes [0.4 mM of ethylene diamine tetraacetic acid (EDTA), 20 mM of Tris-HCl, and 0.4% Triton X-100] prior to centrifugation at 13,000 g for 5 minutes at 4°C. The upper aqueous phase was clearly harvested, and DNA was precipitated overnight by adding an equal volume of isopropanol after adjusting to 0.5 M of NaCl. DNA was again harvested by centrifugation for 10 minutes at 12,000 rpm, and the supernatant was decanted. The pellet was washed twice with 1 mL of 70% ethanol, followed by centrifugation. Dried briefly, the DNA was resuspended in 40 μL of TE buffer and 15-μL were electrophoresed on a 1% agarose gel and stained with 0.5-μg/mL ethidium bromides for visualization.
Isolation of RNA and reverse-transcription polymerase chain reaction (RT-PCR)
The equal amount of HepG2 cells were cultured for 48 hours at various doses of AE, as mentioned above. Total cellular RNA of each treatment was extracted by using Brizol Reagent (Bioflux), which was reverse-transcribed into cDNA by using 2 μg of total RNA, according to the manufacturer's protocol (RNA PCR kit M-MLV, 200 U/mL; Promega). PCR, with 4 pmol of each primer, 1 U of TaqE (Promega), and 1 μL of cDNA, was performed for 5 minutes at 94°C, then 30 cycles of 30 seconds at 94°C, 40 seconds at 58°C, and 30 seconds at 72°C. After the last cycle, there was a final extension for 7 minutes at 72°C. The PCR primers for β-actin, Bcl-2, and vascular endothelial growth factor (VEGF) were as previously reported. 18 –20 Bands of PCR-amplified products were scanned and analyzed semiquantitatively, using an imaging system (Gel Doc EQ; Bio-Rad, Madrid, Spain). All the experiments were repeated at least three times, and similar results were obtained. The mRNA levels of the studied genes were normalized versus those for β-actin.
Immunocytochemical analysis of Bcl-2, VEGF, and p-STAT3
The expressions of Bcl-2, VEGF, and p-STAT3 in AE-treated HepG2 cells were analyzed immunocytochemically. Exponentially growing cells were collected and 4 × 104 cells in medium were pipetted onto coverslips, which had been put in 6-well microplates in advance. After overnight incubation, different concentrations of AE (ultimate concentrations were 25, 50, 100, and 200 μg/mL, respectively) were added and then were incubated again at 37°C in a 5% CO2 atmosphere for 48 hours. The coverslips with cells growing were taken out and washed thrice with PBS (pH = 7.4). The cells were fixed with 80% precold acetone for 15 minutes. Endogenous peroxidase was blocked with 3% (v/v) H2O2 for 5–10 minutes at room temperature. The coverslips were washed three times for 5 minutes with 0.1 M of PBS (pH = 7.4) and incubated for 30 minutes at 25°C in a protein-blocking solution containing 5% bovine serum albumin (BSA). After discarding extra blocking solution, the primary antibody, rabbit antihuman VEGF, bcl-2, c-fos, and p-STAT3 antibody (1:200) were applied to the coverslips for 1 hour at 37°C in a moist chamber. Then, the residual antibody was washed off with 0.1 M of PBS (pH = 7.4; three times, 5 minutes each time) and allowed to react with the biotinylated secondary antibody, namely, biotin-goat antirabbit IgG for 30 minutes. After three 5-minutes washes with PBS, cells were treated with streptavidin-peroxidase for 20 minutes and then incubated with 3,3-diaminobenzidine solution for 5–10 minutes after washing. Finally, the reaction was stopped by washing with distilled water. The coverslips were fixed on glass slides with arabic gum. The slides were examined by using a light microscope at × 200 magnifications. The average intensity and positive area percentage of the slices were determined from using Image-Pro-Plus 6.0 analysis software.
Results
Effect of AE on proliferation ability of HepG2 cells
MTT assay showed that the herbal extract had significant inhibitory effects on the proliferation of HepG2 cells (Table 1). Inhibition of proliferation was observed as early as the 24 hours when cells were treated with AE at a concentration of 100 or 200 μg/mL. Further, 48 hours treatment by AE showed a potent inhibitory effect on HepG2 cells in a dose-dependent manner (P < 0.01).
Mean ± Standard deviation.
P < 0.05; **P < 0.01 versus control.
Effect of AE on cell cycle of HepG2 cells
Cell-cycle distribution of HepG2 cells was determined by flow cytometry. As shown in Table 2, AE could not significantly change HepG2 cell distribution in the G0/G1, S, and G2/M phases after treatment for 48 hours.
Effect of AE on HepG2 cell apoptotic induction
To confirm the induction of cell apoptosis by AE, an Annexin V–FITC/PI assay, based on flow cytometry, was used to examine the early apoptotic cells. Cells undergoing apoptosis would first express phosphatidylserine on the outer leaflet of the cell membrane, which was marked by Annexin V–FITC binding, and followed by the membrane becoming compromised, which was marked by PI intercalation in the cellular DNA and RNA. As shown in Figure 1, the lower left quadrant shows viable cells. The lower right-hand cells are early apoptotic cells, while those in the upper right quadrant are late-stage apoptotic or necrotic cells. As shown in Table 3, only 2.48% ± 0.27% of the control cells were in the early stages of apoptotic induction. However, from incubation with AE for 48 hours at the indicated concentrations, with a concomitant increase in drug concentration, the early apoptotic population rose to 4.86% ± 0.45%, 8.15% ± 1.37%, 12.57% ± 0.76%, and 14.87% ± 0.75%, respectively, suggesting that these cells were expressing phosphatidylserine on the outer leaflet of the membrane, and that these cells might be undergoing apoptosis.
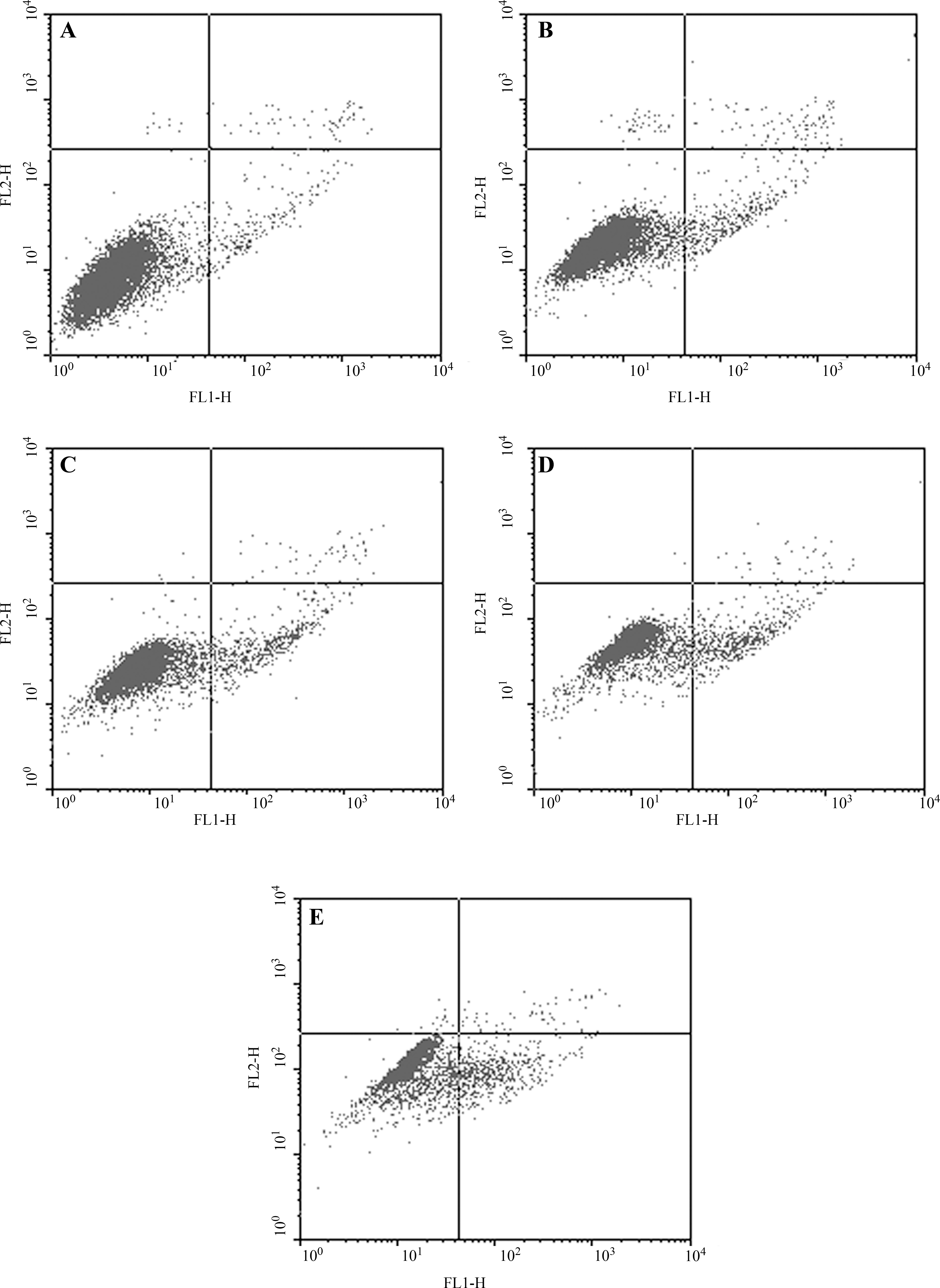
Annexin V and propidium iodide staining of HepG2 cells exposed to various concentrations of AE. The x-axis shows Annexin V–FITC binding, and the y-axis is the staining of propidium iodide. (
P < 0.01; ***P < 0.001 versus control.
Effect of AE on DNA fragmentation
DNA fragmentation is a typical biochemical feature of apoptosis. To further confirm the occurrence of AE-induced apoptosis, a concentration-dependent cellular DNA fragmentation was observed by gel electrophoresis, and a ladder-like pattern, a typical character of DNA cleavage between nucleosomes, was visible after incubation with 25, 50, 100, and 200 μg/mL of AE for 48 hours (Fig. 2). Also, the intensity of banding was more prominent at the highest dose, compared to the others.
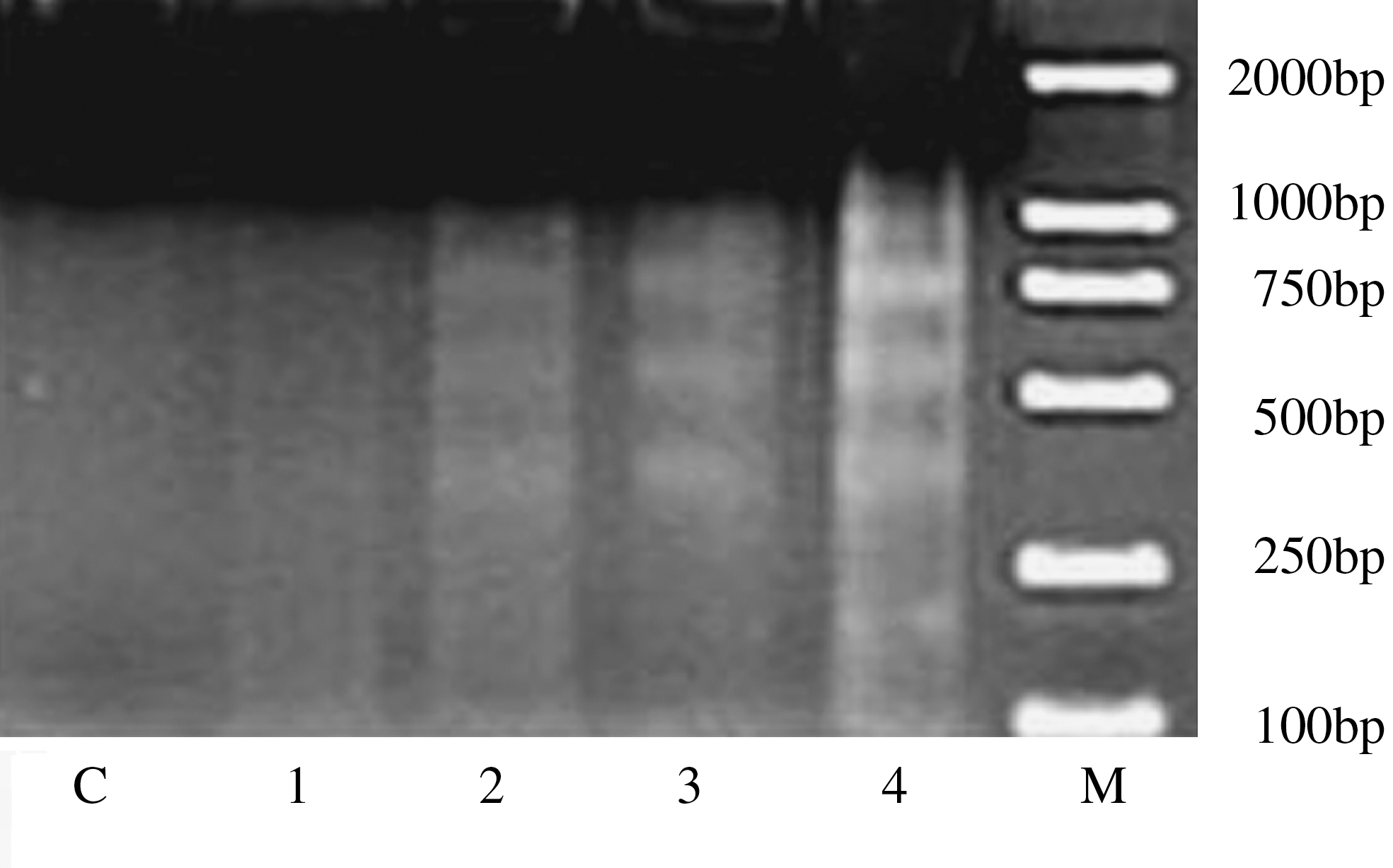
DNA fragmentation of control or treated HepG2 cells by AE was measured by ethidium bromide staining. C, control; M, (DNA marker); lanes 1–4, the effects of 48-hours exposures to 25, 50, 100, and 200 μg/mL, respectively.
Effect of AE on Bcl-2 and VEGF mRNA levels
The expression of Bcl-2 and VEGF genes after drug treatment was examined by means of RT-PCR analysis. The RT-PCR products were run in a 1.5% agarose gel and scanned by using a densitometer. The product sizes of bcl-2, VEGF, and β-actin (housekeeping gene) were 124, 212, and 389 bp, respectively (Fig. 3A). All data are expressed as the ratio of the densities of target genes to that of β-actin. As shown in Figure 3B, after 48 hours of incubation with AE, the mRNA expression level of Bcl-2 was significantly decreased at 50 (P < 0.05), 100 (P < 0.05), and 200 μg/mL (P < 0.001); AE also significantly declined the mRNA level of VEGF at 25 (P < 0.05), 50 (P < 0.05), 100 (P < 0.05), and 200 μg/mL (P < 0.05).
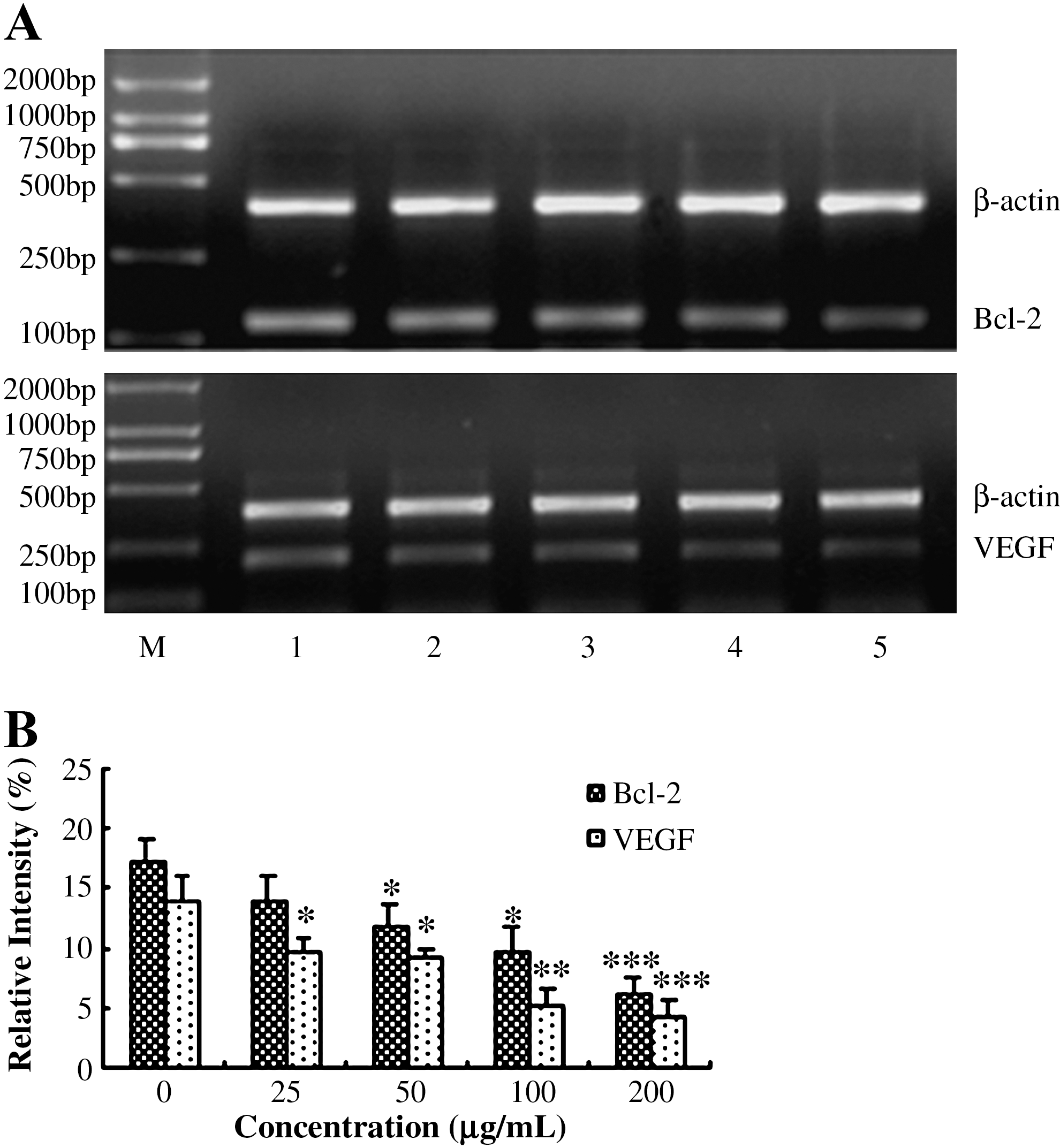
Expressions of Bcl-2 and VEGF mRNA in HepG2 cells exposed to various doses of AE was analyzed by RT-PCR. (
Effect of AE on protein levels of Bcl-2, VEGF, and p-STAT3
Immunocytochemistry was used to determine the protein levels of Bcl-2, VEGF, and p-STAT3. A positive reaction was characterized by dark-brown staining in the nucleus and/or cytoplasm (Fig. 4). Quantitative measurement of these proteins was carried out by detecting the average intensity and positive area percentage in the slices. Average intensity and positive area percentage indicate the intensity and quantity of gene expression, respectively. The higher average intensity represents higher protein expression intensity. Higher positive area percentage indicates that there are more immunoreactive positive cells in the slices. The image analysis results are shown in Table 4. AE significantly decreased the the average intensity and positive area percentage of Bcl-2 and VEGF at 50, 100, and 200 μg/mL (P < 0.05 or P < 0.01 or P < 0.001). Moreover, AE at all the four doses significantly decreased both the average intensity and positive area percentage of p-STAT3 protein in HepG2 cells in a dose-dependent manner (P < 0.05 or P < 0.01 or P < 0.001). This indicated that these three protein levels of HepG2 were significantly decreased after incubating with AE for 48 hours.

Immunocytochemical detection of VEGF, Bcl-2, and p-STAT3 in HepG2 cells. (
P < 0.05; **P < 0.01; ***P < 0.001 versus control (n = 10).
VEGF, vascular endothelial growth factor; AE, aqueous extract.
Discussion
Although S. sarmentosum is used in the treatment of chronic viral hepatitis in China, in the present study, from using the MTT assay, we found that its AEs exhibited the strongest inhibitory effect on human hepatoma cell proliferation. Further biochemical analysis indicated that the proliferation inhibitory activity of AE was related to the induction of apoptosis in the HepG2 cells after exposure to AE for 48 hours. The characteristic occurrence in the early apoptotic cells is the translocation of phosphatidylserine from the inner leaflet of the plasma membrane to the cell surface. 21 In our studies, the apoptosis evoked by AE was confirmed by the observation of phosphatidylserine translocation. In addition, the dose-dependent formation of DNA fragment was also determined by means of AGE.
As the ability to induce tumor cell apoptosis is an important property of a candidate anticancer drug, and much effort has been directed toward the search for compounds that affect apoptosis and understanding their mechanisms of action, one of the aims of this study was to determine whether possible changes in the expression of Bcl-2 could account for AE-induced cell apoptosis. In particular, the Bcl-2 family members, including pro- (such as Bax) or antiapoptotic (such as Bcl-2 and BclxL) proteins, have been proposed to be critical in the regulation of cellular homeostasis. 22 Using immunocytochemistry, the present results suggest that the levels of Bcl-2 oncoprotein in HepG2 cells were downmodulated by treatment with AE, in comparison to the control group.
Results from the present study suggest that AE downregulates the protein expression of VEGF, which may be responsible for the observed antiproliferation effect of AE on the HepG2 cells. It is well known that vascularization has an important role in tumor growth, invasion, and metastasis because tumor blood vessels not only provide nutrition for tumor growth, but also provide pathways for the pervasion of tumor cells. 23 Both RT-PCR and immunocytochemistry results indicated that the aqueous extract from S. sarmentosum could dramatically downregulate the expression of VEGF.
Further, a significant decrease in protein pSTAT3 was also observed by immunocytochemistry. It is reported that the constitutive activation of STAT3 by tyrosine phosphorylation, in response to cytokines and growth factors, has been demonstrated in several human cancers, 24 –30 including in the hepatoma cells. 31 In fact, STAT3 has been observed to upregulate genes encoding apoptosis inhibitors (Bcl-xL, Mcl-1, and survivin) 32 and angiogenesis inducers (e.g., VEGF), 33 which are important for tumor progression.
Conclusions
Based on the above results, we could make a conclusion that the AE of S. sarmentosum possesses antiproliferation activity on HepG2 cells by inducing cells' apoptosis and downregulating the expression of Bcl-2, VEGF, and pSTAT3, which, probably, is the different mechanism from that of methanol extract. 6 However, further investigations at the molecular level are required to identify the signal-transduction pathway by which the apoptosis of HepG2 cells caused by AE occurs.
Footnotes
Acknowledgments
This study was financed by the National Nature Science Foundation of China (no. 30873188 to WYZ).
Disclosure Statement
No competing financial interests exist.