
Abstract
Nonviral delivery systems are relatively easy to produce in the large scale, are safe, and elicit a negligible immune response. Nanoparticles (NPs) offer promise as nonviral vectors as biocompatible and -degradable carriers of drugs with targeting to specific sites by surface receptors of monoclonal antibodies (mAbs). We investigated the effect of four PEG-PLGA (polyethylene glycol-polylactic-co-glycolic acid) NP systems on drug-resistant B-chronic lymphocytic leukemia (B-CLL) cells in vitro, three of them encapsulating the drug, hydroxylchloroquine (HDQ), two with NP surface coatings of mAbs (NP1) CD20, (NP2) CD19, and CD20, and one (NP3) with no mAb, but tagged with the fluorescent marker, fluorescein isothiocyanate. The fourth NP system (NP4) was coated with anti-CD19/FITC and anti-CD20/Alexa-Fluor® antibodies, but did not contain the active drug, HCQ. Our data indicate that PEG-PLGA nanoparticles with surface mAbs are suitable for selective drug delivery to B-CLL cells and produce a strong apoptotic effect when loaded with the lysosomotropic agent, HDQ.
Introduction
Despite many advances and new discoveries in the field of leukemia, B-chronic lymphocytic leukemia (B-CLL) continues to be an incurable progressive clonal disease. 1 Either prolonging survival or developing new therapeutic effective modalities are still difficult aims to be attained. 2 Along these lines, agents with novel mechanisms of action as well as new approaches for antileukemic drug delivery are crucial to overcoming multidrug resistance in B-CLL. 3 –5 In general, the standard initial therapy has been single-agent chemotherapy typically employing the drug, chlorambucil (CLB). 6 Recently, fludarabine has been incorporated as the front-line treatment for B-CLL. 7 Various monoclonal antibodies (mAbs) such as anti-CD20 (rituximab and ofatumumab), 8,9 anti-CD52 (alemtuzumab), 10 and combinations of these antibodies, 11 with many others, 12 have shown promise for addressing B-CLL treatment. At least 60%–80% of patients usually respond to these therapies, and often for years, but eventually, all patients finally become resistant to these agents. 4 Upon failure of these therapies, the patients do not achieve complete remission, and they relapse or fail to respond to almost all available therapeutic agents, 13 which carries high, and often unacceptable, acute and chronic organ toxicity with an increased risk for secondary malignancies. 14 Traditional, anti-CLL agents, such as CLB, are transported by carrier-mediated systems into cells and generally alkylate to DNA, RNA, and proteins. 6 The precise mechanisms responsible for the development of drug resistance in the clinical setting are not known. 13 Ordinarily, CLB-resistant B-CLL lymphocytes are 5- to 6-fold more resistant in vitro, using the MTT assay, as compared to sensitive lymphocytes (IC50 CLB of ≤61.0 μmol/L can be considered sensitive). 15 Various factors have been related to drug-resistance development, poor treatment outcome, and aggressive B-CLL response, including, p53 gene mutations, abnormal angiogenesis, overexpression of bcl-2, and a complex altered repertoire of cytokines and growth factors. 16 In 2001, Lagneaux et al. 17 performed in vitro testing by using hydroxychloroquine (HCQ), an antirheumatic drug, on malignant B-cells, and results showed early induction of apoptosis with HCQ by activation of caspase-3 and no protection by survival factors, such as in bone marrow stromal cells. The mean IC50 was 32 ± 7 and >100 μg/mL at 24 hours of exposure for CLL cells and normal lymphocytes, respectively. 17,18 However, these levels of drug concentration are very difficult or impossible to reach in vivo with common oral or parenteral administration. 19 It is clear, then, that the use of traditional or new drugs with novel mechanisms for the treatment of B-CLL would benefit from advanced therapeutic strategies. In this way, it could be possible to obtain synergistic effects in order to overcome pharmacokinetic obstacles or specifically deliver effective amounts of drugs straight into the targeted B-CLL cells, improving the overall survival and decreasing treatment-associated morbidity. We report in this article an innovative technology, based on biodegradable nanoparticles, that could avoid these problems and introduce a new way of treatment modality, especially for drug resistant B-CLL. The aim of this work was to investigate the in vitro apoptotic effect of HCQ-loaded biodegradable nanoparticles with surface coatings of different monoclonal antibodies on drug resistant B-CLL cells.
Methods
Chemicals used in this study were reagent grade or better. Some of the chemicals purchased were polyethylene glycol (Nektar, San Carlos, CA), HCQ sulfate (ACROS, Geel, Belgium), human mAbs CD19 and CD20 (BioLegend, San Diego, CA), a B-Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany), AIM V serum-free medium (GIBCO/Invitrogen, Carlsbad, CA), and ethidium bromide/acridine orange staining (FluoroQuench; One Lambda, Canoga Park, CA). Biodegradable nanoparticles (BNPs) of polylactic-co-glycolic acid (PLGA) were purchased from BioTarget, Inc. (Chicago, IL), with effective diameters ranging from 200 to 300 nm, based upon dynamic light scattering (DLS). The particles were produced under class 100 clean room conditions by a proprietary emulsion process.
BNPs
Four different types of BNPs were used for testing (Fig. 1). The therapeutic agent, HCQ sulfate (ACROS), was encapsulated at a concentration of 165 μg per mg of polymer inside three of the four types of BNPs. Two types of HCQ-BNPs (HCP-encapsulated BNPs) were surface modified with mAbs of either antihuman CD19 (designated BNP1), or antihuman CD19 and antihuman CD20 (designated BNP2). The anti-CD19 and CD20 mAb concentration was 5.75 and 5.38 μg, respectively, per mg of polymer. A fluorescent marker was attached to each mAb; Fluorescein isothiocyanate (FITC)-conjugated anti-CD19 (fluorescein isothiocyanate exhibits excitation at 495 nm and emission at 525 nm) and Alexa-Fluor®–conjugated anti-CD20 (excitation at 696 nm and emission at 719 nm) (Table 1). The mean number of molecules per particle in the BNP surface was 959 for CD19 and 366 for CD20. An additional HCQ-BNP system was produced with a fluorescent polymer (PEG-FITC) and no antibody (designated BNP3). The fourth BNPs system was coated with anti-CD19/FITC and anti-CD20/Alexa-Fluor antibodies, but did not contain the active drug, HCQ (designated BNP4). All BNPs were suspended at time of use in phosphate-buffered saline (PBS) with 10% bovine serum albumin (BSA) at a final total mass concentration of 900 μg/mL. Aliquots of 0, 0.1, 0.2, 0.5, 1, and 2 μL from these solutions were used for all experiments.
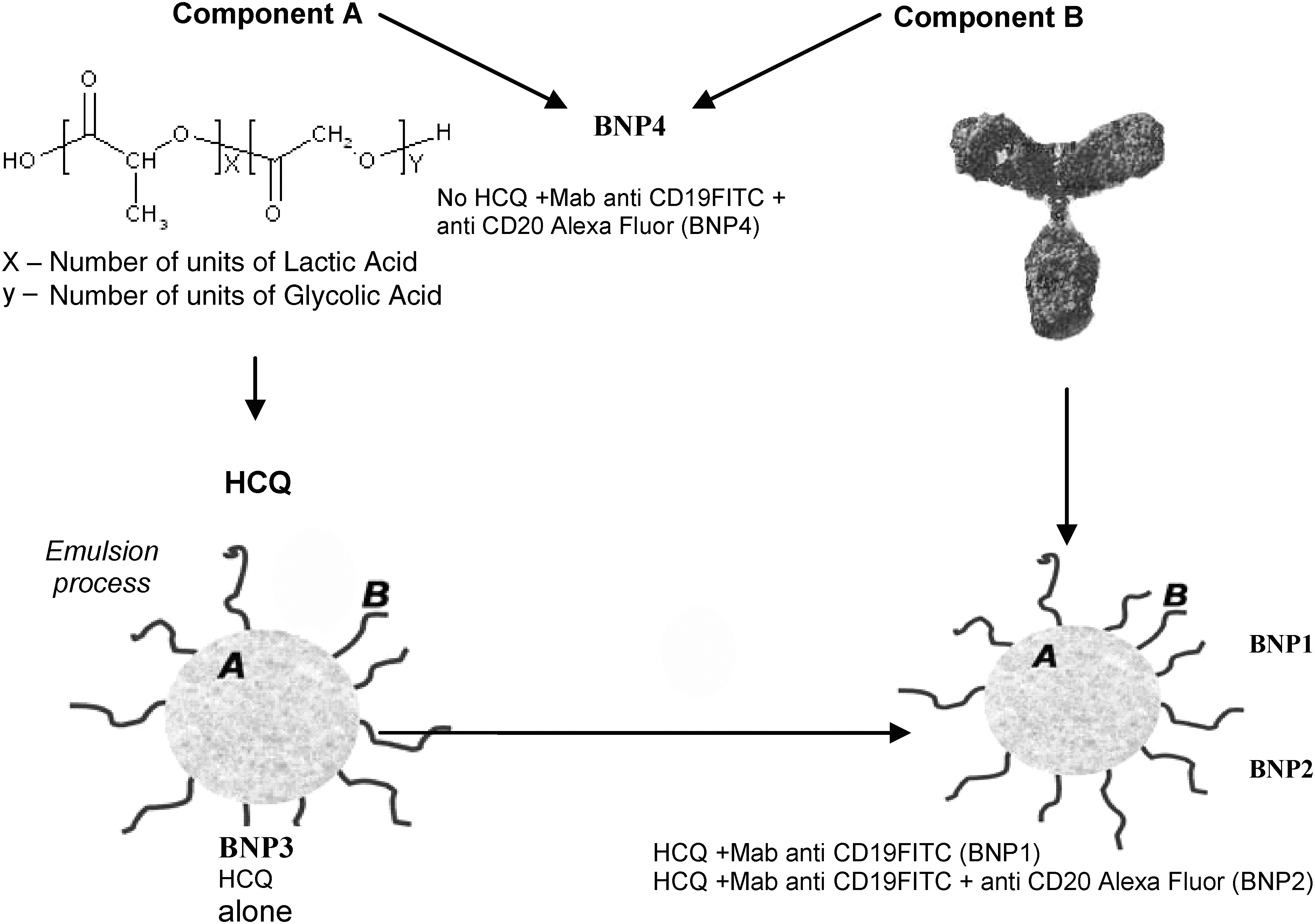
Targeted biodegradable nanoparticle design scheme.
FITC, Fluorescein isothiocyanate; MAb, monoclonal antibody; HCQ, hydroxychloroguine.
B-CLL cells and normal B-lymphocytes
Blood samples were obtained after informed consent from healthy volunteers and from B-CLL patients (Rai-IV, heavily pre-treated and resistant to chlorambucil and fludarabine). The mononuclear cell fractions were isolated by centrifugation on Ficoll-Hypaque gradients. Normal B-cell and primary tumor B-cells (positive for CD5, CD19, CD23, CD20, CD38, and ZAP70, with unmutated Ig genes and p53 mutations) were isolated from normal and B-CLL samples, using the B-Cell Isolation Kit (Miltenyi BioTec). Isolated cells were purified at greater than 95% with fewer than 2% T-lymphocytes, as determined by flow cytometry with anti-CD3. Double staining with anti-CD19 and sIg showed that normal B-cells were present at less than 5% in the leukemic samples. When the B-CLL and normal B-lymphocytes were tested for in vitro drug resistance, the LD50 of CLB in the lymphocytes from the CLL patient was 6-fold greater than the average LD50 of the lymphocytes from the normal subject.
Cell culture with BNPs
The freshly isolated cells were incubated for all experiments at 37°C and 5% CO2 at various concentrations of BNPs in AIM V® serum-free medium. Briefly, 4 × 105 cells, both from the CLL patient and the healthy control, were incubated in triplicate in 0.5 mL of AIM V® serum-free medium for all experiments. The cells were cultured in plastic sterile tubes in the presence of 0, 0.1, 0.2, 0.5, 1, or 2 μL of each BNP system solution, as well as with a combination of FITC-conjugated anti-CD19 and Alexa-Fluor–conjugated anti-CD20 antibodies suspended at time of use in PBS with 10% BSA alone without BNPs. After 15 minutes of incubation, cell aliquots were taken and were observed by confocal microscopy. After 4 hours of incubation, samples were washed three times with PBS and aliquots were taken for confocal microscopy observation, followed by additional incubation for a total of 44 additional hours in AIM V serum-free medium.
Confocal microscopy and fluorescence correlation spectroscopy
Confocal Imaging combined with fluorescence correlation spectroscopy (FCS) was performed on all our BNP systems and BNPs after incubation with cells at 15 minutes and 4 hours. All FCS measurements were acquired from using a standard Alba FCS spectrometer (ISS, Champaign, IL) and a Nikon microscope (Model TE2000-E), equipped with a water-immersion (NA = 1.2, 60X) objective. Acquisition of the fluorescence by filters allowed the separation of two bands: 500–550 nm (channel one) and 700–750 nm (channel two). Two cooled avalanche photodiodes (APDs) were utilized as light detectors in photon-counting mode. The software utilized for data acquisition and analysis was the Vista package by ISS. The observation volume of the instrument, using a 50-μm-diameter pinhole, was determined prior to measurements from using a solution of rhodamine 6G, using the 473 nm; the axial resolution was determined as 0.25 μm and the z-resolution as 1.1 μm. Measurements where acquired from using the 50-μm-diameter pinhole. The autocorrelation function was built by dividing the observation time scale in intervals, measuring the number of photons collected in each time interval. The autocorrelation function was fitted with a physical model describing the sample. The results were the diffusion coefficient and the number of molecules per unit volume within the observation volume. Based on the specific lifetime, diffusion constants for particles could be determined. All cell samples were also observed with an Olympus laser confocal fluorescence microscope, evaluated for BNP cell penetration, and photographed with an Olympus digital camera after the first 4-hour incubation period and washing steps. Images were analyzed with acquisition FluoView software, version 3.3. For the confocal observations, laser excitations were at 525 nm (green) and 719 nm (red), and the cross correlation was also provided. Further, DAPI (4',6-diamidino-2-phenylindole) nuclear staining of the cells, cultured with the different BNPs, was done and observed in blue for laser excitation at 461 nm.
Cell viability testing
We analyzed the percentage of living and dead cells in each sample at 0, 24, and 48 hours of incubation with each of the BNP systems by using ethidium bromide/acridine orange staining and immune-fluorescence microscopy counting.
Zeta potential and DLS
Nanoparticle size-distribution and zeta-potential measurements of all four BNP systems were performed on a ZetaPlus Analyzer (Brookhaven Instruments, Corp.). Particle-size calibration was performed with a 100-nm NIST (National Institute of standards and technology, Gaithersburg, MD) polystyrene latex standard, and the zeta potential was verified with a proprietary standard (BI-ZR3) provided from the manufacturer. Adjustments to pH were done with dilute NaOH or HNO3 solutions.
Fluorescence-activated cell (FACS) apoptosis analysis
Measurement of cell apoptosis, using Annexin-V and propidium-iodide by FACS analysis, was made at 0, 24, and 48 hours of incubation with each type of the BNP systems.
Statistical analysis
The statistical significance of differences among groups was assessed by using the Student's t-test with a 5% limit for statistical significance. Correlation analysis was performed by using a regression test.
Results
FCS analysis provided the lifetimes of the anti-CD19 and CD20 markers coated on the BNPs. These markers had distinguishable lifetimes, which were monitored spatially and temporally. The spatial characteristics of the anti-CD19 and the anti-CD19/anti-CD20 BNPs demonstrated rapid cell internalization for both systems after 15 minutes of incubation (Fig. 2a), with anti-CD19 having more rapid internalization than the anti-CD20. In contrast, the anti-CD20 mAb seemed to preferentially establish surface-cell interactions after 15 minutes of incubation (Fig. 2b). FCS also provided independent evidence for the median diameter (299 nm) by the diffusion coefficients of the different BNP systems used. The FCS-calculated diameter was nearly identical to the effective median diameter of 310 nm obtained from laser-light scattering experiments in our BNP systems. BNPs formulated with mAbs resulted in significant penetration after 4 hours of incubation in both normal lymphocytes and B-CLL cells, compared to BNPs without antibodies (P < 0.001) (Fig. 3). The mean cell viability of normal and leukaemic cells samples prior to apoptosis experiments was 96.4% and 95.3%, respectively. All HCQ-loaded anti-CD19- and anti-CD19/anti-CD20–coated BNPs efficiently induced apoptosis of malignant human B-CLL cells in vitro. At the 0.5-μL concentration, HCQ-antiCD19-BNPs induced 4-fold more apoptosis in malignant B-CLL cells, compared to normal lymphocytes (P < 0.001). When BNPs were simultaneously coated with both antibodies and injected at the same concentration (0.5 μL), the most effective apoptotic effect was over 5-fold more pronounced on B-CLL cells, compared to normal lymphocytes (Fig. 4). Reductions of living B-CLL cells were observed in vitro at 24 and 48 hours for injections of all concentrations of mAbs coated nanoparticles loaded with HCQ. Loss of viability in human CLL cells correlated with the early induction of apoptosis. HCQ-BNPs with mAbs induced a decrease in cell viability in a dose- and time-dependent manner. In leukemic cells, the nanoparticles reduced cell viability in doses and times significantly lower than in normal lymphocytes (Fig. 5). In vitro treatment of drug-resistant B-CLL cells with these HCQ-loaded BNPs (BNP1, BNP2, and BNP3) was shown to be significantly more effective (P < 0.001) than BNPs without drug (BNP4), indicating that treatment with empty BNPs had little impact on cell viability. BNPs encapsulated with HCQ, but without mAbs (BNP3), had significantly less impact on in vitro cell viability (P < 0.001) (Fig. 5). FITC-conjugated anti-CD19 and Alexa-Fluor–conjugated anti-CD20 antibodies suspended in PBS with 10% BSA in AIM V medium alone without BNPs produced no significant apoptotic effect in B-CLL or normal lymphocytes.

Fluorescence correlation spectroscopy micrograph of anti-CD19 (

Penetration of nanoparticles into normal and B-CLL after 4 hours of incubation. BNP1 (nanoparticles are surface coated with anti-CD19 monoclonal antibodies and loaded with the therapeutic agent, HCQ); BNP2 (nanoparticles are surface coated with anti-CD19/anti-CD20 monoclonal antibodies and loaded with the therapeutic agent, HCQ); BNP3 (HCQ with fluorescent polymer—PEG/FITC—and no antibody). Assays n = 9 for each BNP and each type of cells. Differences between BNP1 or BNP2 and BNP3: P < 0.001.

Cellular apoptosis induced by BNP2 nanoparticles as a function of BNP2 concentration in normal and B-CLL cells (BNP at .5-uL concentration; incubation time = 24 hours). BNP2 nanoparticles are surface coated with anti-CD19/anti-CD20 monoclonal antibodies and loaded with the therapeutic agent, HCQ.
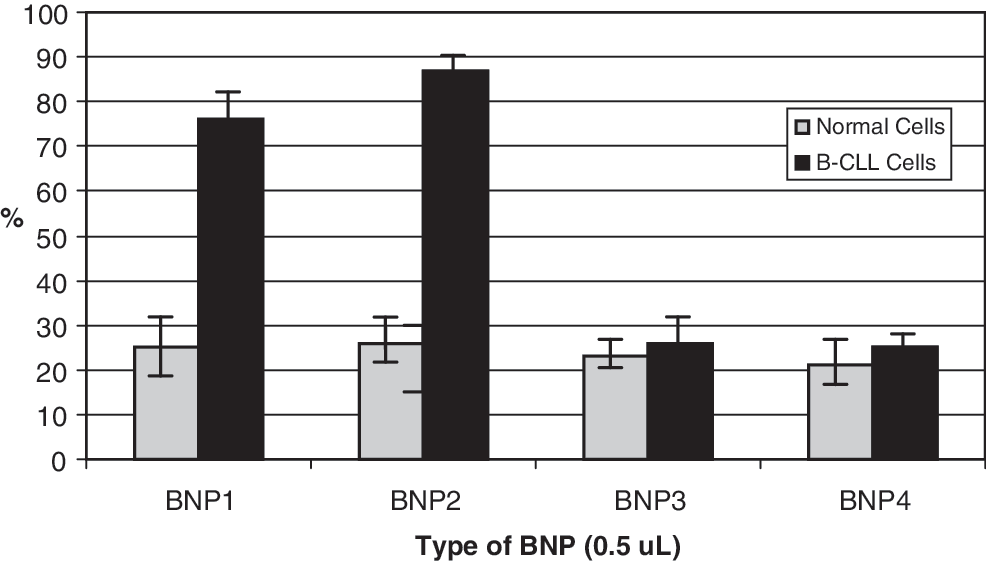
Cellular mortality of B-CLL and normal cells induced by nanoparticles after 48 hours of incubation. BNP1 (nanoparticles are surface coated with anti-CD19 monoclonal antibodies and loaded with the therapeutic agent, HCQ); BNP2 (nanoparticles are surface coated with anti-CD19/anti-CD20 monoclonal antibodies and loaded with the therapeutic agent, HCQ); BNP3 (HCQ with fluorescent polymer—PEG/FITC—(and no antibody); BNP4 system coated with anti-CD19/FITC and anti-CD20/Alexa-Fluor antibodies without HCQ. Assays n = 9 for each BNP and each type of cells. Differences between leukaemic cell submitted to BNP1 or BNP2 and BNP3 or BNP4: P < 0.001.
Discussion
In clinical oncology, especially for the treatment of CLL, the development of new anticancer agents with different modes of action is of key importance for overcoming therapy resistance. The antimalarial quinolines, chloroquine, hydroxychloroquine, and quinidine, are defined as antiproliferative compounds in the National Cancer Institute database. 20 These drugs can stimulate cell differentiation at growth-inhibitory concentrations as well as inhibition of histone deacetylase (HDAC) activity in vitro, DNA damage, and apoptosis of some tumor lines. 21 HCQ exhibits a pharmacologic effect by increasing the pH of intracellular organelles, including the lysosome, 22 endosome, and the trans-Golgi network, inducing lysosomal membrane permeabilization (LMP), 23 and, perhaps, activating p53-independent apoptosis. 24 Lysosomotropic agents, such as HCQ, induce LMP, which is known to cause a translocation of lysosomal proteases to the cytosol. The lysosomal cell-death pathway has been suggested to represent a potential cancer treatment, but anticancer drugs that trigger this pathway are not in clinical use. 22 Considering the high frequency of p53 mutations in human cancer cells, the demonstration that LMP is potentially a common mechanism of action of p53-independent drugs is interesting in terms of future drug development. 23 HCQ is usually extremely well tolerated and is not associated with too many side-effects (aside from some degree of retinal and ototoxicity). 24 Pharmacokinetic analyses with HCQ demonstrated large inter- and intrapatient variability. Maximum HCQ blood concentration in healthy volunteers receiving an 800-mg dose of hydroxychloroquine sulphate orally ranged from 135 to 422 ng mL−1 and time to maximum (tmax) was 1.5–7.0 hours, showing significant differences between subjects. Half-life and mean residence time for HCQ were long (around 40 days), and large volumes of distribution were calculated (5522 L from blood, 44,257 L from plasma). Sequestration into the tissues is an important feature of the disposition of HCQ. The persistence of HCQ in the body is due primarily to this extensive tissue distribution, rather than to low clearance. In this way, concentrations selectively high enough on B-CLL lymphocytes to cause effective apoptosis as seen in vitro are impossible to reach in the in vivo setting by oral administration of HCQ. 25 In order to use the antineoplastic advantages of this kind of drug, and to deliver high levels of HCQ selectively inside B-CLL cells, a new pharmacologic delivery strategy, such as the one shown in this article, has been designed. Protein expression values for CD19 and CD20 in leukemic B-cells can differ from those of normal blood B-lymphocytes and provide promising targets for attachment and intracellular entry into B-CLL cells. 26 Analysis of HCQ loaded/antibody-coated BNPs effects on B-CLL cells clearly showed that these particles are potent inducers of tumor cell death for in vitro applications. Further, HCQ-loaded BNPs efficiently induced CLL cell apoptosis with little effect on the viability of normal B-cells in vivo at the same concentrations. In this way, the delivery of B-CLL-specific, proapoptotic drugs such as HCQ inside the cytoplasm of B-CLL malignant cells is possible from using biodegradable nanoparticles by targeting surface markers, such as CD19 and CD20. These HCQ-loaded, mAb-surface–modified nanoparticles allow for specificity in the treatment of B-CLL cells without harming normal lymphocytes, thus overcoming pharmacokinetics difficulties found in the past. It is also possible to deliver additional anticancer drugs, such as chlorambucil, inside this type of BNPs with or without one of the antimalarial drugs (e.g., HCQ, chloroquine, primaquine, or many others) and perhaps enhance the effect of chlorambucil. Since chlorambucil has been one of the most used and well-tolerated drugs in CLL therapy, the combination of this drug with the present nanoparticles with or without HCQ would be of great interest for further studies. It is also planned, to test as lead clinical candidates, BNP systems with only antihuman CD20 antibody (e.g., rituximab or ofatumumab) or other mAbs, such as anti-CD52 (e.g., alentuzumab), anti-CD23 (e.g., lumiliximab), anti-CD38, and/or anti-CD21/CD22, 27,28 as well as other innovative anti-CLL drugs, such as those from natural origin and their combinations in the same nanoparticle.
Conclusions
The data presented in this article suggests that HCQ-BNPs trigger a selective, apoptotic response in B-CLL cells at low concentrations when specific mAbs, such as anti-CD19 and/or anti-CD20, are coated on their surface and that apoptosis shifts to necrosis at higher concentrations. Our system offers a new, powerful, and less-toxic therapy for B-CLL and other B-cell malignancies. Further, a treatment option that combines a therapeutic drug, such as HCQ or other drugs of natural origin, with the appropriate mAb-coated nanoparticle may extend the application of this approach to other conditions in which the elimination of a specific cell population is desired, such as in autoimmune diseases.
Footnotes
Disclosure Statement
No competing financial interests exist.