
Abstract
Aim:
Insulin-like growth factor-I receptor (IGF-IR) is widely overexpressed in a variety of human cancers including ovarian cancer. It plays an important role in cancer cell proliferation and tumor growth. In this study, small interfering RNA (siRNA) was used to silence IGF-IR gene expression in the human ovarian cancer cell line OVCAR3 and then its effects on proliferation, apoptosis, angiogenesis, and the growth of tumor cells in vitro and in nude mice were evaluated.
Methods:
Three siRNA sequences for the IGF-IR gene were cloned into expression plasmids and transfected into OVCAR3 cells. The downregulation of IGF-IR expression at both mRNA and protein levels were detected by real-time polymerase chain reaction and western blot analysis. Cell proliferation inhibition rates were determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Nude mice (n = 6 per group) were subcutaneously xenografted with 2 × 106 OVCAR3 cancer cells. Tumor growth, cellular proliferation (Ki-67 immunohistochemistry), apoptosis, and angiogenesis (CD31 immunohistochemistry) were compared for mice administered either IGF-IR–specific or negative control siRNA over 5 weeks.
Results:
The mRNA and protein expression of IGF-IR was significantly decreased at 48 hours after transfection, leading to a potent suppression of tumor cell proliferation in vitro. The IGF-IR–specific siRNA dramatically suppressed tumor growth and cellular proliferation, as well as promoted tumor cellular apoptosis and inhibited angiogenesis in an OVCAR3 s.c. xenograft model.
Conclusions:
The siRNA targeting of IGF-IR can effectively inhibit the growth of ovarian cancer cells and may be used as a potent therapy.
Introduction
Epithelial ovarian cancer is one of the most lethal gynecologic malignancies. This tumor is generally asymptomatic and spread widely into pelvic and abdominal cavities at the time of diagnosis. Current management for advanced epithelial ovarian cancer is cytoreductive surgery followed by combination chemotherapy. First-line chemotherapy with platinum drugs and taxanes yields response rates of over 80% in these cases, but almost all patients will relapse, highlighting the need for effective and well-tolerated regimens.
Mounting evidence suggests that both insulin-like growth factors (IGF-I and IGF-II) and their receptors (IGF-IRs) are involved in the development and progression of cancer. 1,2 IGF-IR is perhaps the most prominent factor, and it has been extensively studied for its role in the proliferation and differentiation of cancer cells. 3,4 IGF-IR is a transmembrane heterotetrameric protein, encoded by the IGF-IR gene located on chromosome 15q25–q26. IGF-IR is composed of two α and two β subunits linked by disulfide bonds. The extracellular α subunit is responsible for ligand binding, whereas the β subunit consists of a transmembrane domain and a cytoplasmic tyrosine kinase domain. 5 The binding of its ligands (IGF-I, IGF-II) to the extracellular domains of IGF-IR activates its intracellular tyrosine kinase domain resulting in trans-β subunit autophosphorylation and the stimulation of signaling cascades that include the SHC/Grb2/SOS/Ras/raf/MEK/MAPK and IRS/PI3-K/Akt/p70s6k pathways, leading to a host of effects, including proliferation, tumorigenesis, and inhibition of apoptosis. 3,4,6,7
High levels of expression of IGF-IR have been reported in a broad range of human malignancies, including ovarian cancer. 4,7 –10 In a culture study of rabbit ovarian mesothelial cells, overexpression of IGF-IR was found to induce malignant transformation of the cells. 11 IGF-IR is present in surgical specimens from primary or metastatic ovarian tumors. 9 Cells with IGF-IR deleted are resistant to oncogenic transformation. 3 Therefore, IGF-IR is an attractive antitumor target.
Inhibition of IGF-IR by various approaches, including IGF-IR antisense oligonucleotides, 12 IGF-IR–blocking antibodies, 13 dominant-negative IGF-IR, 14 and small-molecule inhibitors, 15 have been shown to reduce tumor growth in human tumor xenograft models. But the stability and delivery efficacy of these agents seems to be a crucial limiting factor in exerting an inhibitory effect on the targeted molecule.
RNA interference (RNAi) is the sequence-specific, post-transcriptional gene-silencing method initiated by double-stranded RNA that are homologous to the gene being suppressed. 16 This technology is commonly used as a research tool, and its use in clinical settings is now being explored. The high efficiency and specificity of RNA-mediated interference has made it a powerful and widely used tool in cancer therapy. 17
In the present study, small interfering RNA (siRNA) targeting to the IGF-IR gene was introduced into human ovarian cancer cell line OVCAR3, which overexpresses IGF-IR. Its effect on cancer cell growth and its therapeutic efficacy in a xenograft model were investigated.
Materials and Methods
Cell line and cell culture
The human ovarian cancer cell line OVCAR3 was obtained from the Institute of Life Science, Tianjing, China. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone Corp.) supplemented with 10% fetal bovine serum (HyClone Corp.), 105 units/L penicillin, and 0.1 g/L streptomycin at 37°C in a humidified atmosphere containing 5% CO2. These studies were carried out with approval of the Medical Ethical Committee of Harbin Medical University.
Design and synthesis of shRNAs
Three siRNAs targeting human IGF-IR gene (NCBI, accession number NM000875) and one scrambled siRNA (used for a negative control) were used. The sequences are shown in Table 1.
IGF-IR, insulin-like growth factor-I receptor; siRNA, small interfering RNA.
These sequences were selected in accordance with the general guidelines regarding efficacy of siRNA targeting proposed by Elbashir et al. 18 Basic local alignment search tool (BLAST) searches of the human genome database were carried out to ensure the sequences would not target other gene transcripts. The negative control was the siRNA sequence with no homology to any human gene sequence. All siRNA were designed and synthesized by Genesil Biotechnology Co. The recombinant plasmids containing siRNA sequences 1, 2, 3, and negative control were named pSih IGF-IR-1, pSih IGF-IR-2, pSih IGF-IR-3, and pHK respectively.
Transfection of siRNA
OVCAR3 cells were seeded (2 × 105 cells/well), without antibiotics, in six-well tissue culture plates. After 24 hours of incubation, when the cells reached 70%–80% confluence, pSih IGF-IR-1, pSih IGF-IR-2, pSih IGF-IR-3, and pHK were transfected into OVCAR3 cells using Lipofectamine™2000 (Invitrogen Corp.) according to the manufacturer's instructions. After 6 hours at 37°C, the medium was changed, and the cells were cultivated in DMEM supplemented with 10% heat-inactivated fetal bovine serum. The transfections were detected by fluorescence microscopy. Cells transfected with pHK, pSih IGF-IR-1, pSih IGF-IR-2, and pSih IGF-IR-3 were designated as HK, R1, R2, and R3, respectively.
Western blotting of IGF-IR
Cells were collected at 48 hours after transfection, washed three times with ice-cold phosphate-buffered saline (PBS), and then lysed in buffer (50 mmol/L Tris-HCl [pH 8.0], 150 mmol/L NaCl, 100 μg/mL PMSF, and 1% Triton X-100) for 30 minutes on ice. After removal of cell debris by centrifugation at 12,000g for 10 minutes at 4°C, 50 μg of each lysate sample was boiled for 10 minutes in sample buffer, resolved by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and then transferred onto nitrocellulose membrane (Hybond C; Amersham). Membranes were blocked in 5% nonfat dry milk in TBST (10 mmol/L Tris-HCl [pH 7.5], 150 mmol/L NaCl, and 0.1% Tween 20) at room temperature for 1 hour and immunoblotted with mouse anti–IGF-IR antibody (1:1000; Santa Cruz Biotechnology) overnight at 4°C. After three times washing with TBST, membranes were incubated with rabbit anti-mouse antibody linked to horseradish peroxidase (1:10,000; Santa Cruz Biotechnology). To confirm equal protein loading, membranes were reprobed with a 1:1000 dilution of an anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (HaiGene Corp.). Chemiluminescent detection (Santa Cruz Biotechnology) was performed in accordance with the manufacturer's instructions. Protein expression was quantified using Labwork software version 2.0. Blots were performed in triplicate.
Quantitative real-time reverse transcriptase–polymerase chain reaction analysis
Total RNA was extracted from cells using RNeasy kit (HaiGene Corp.) and reverse-transcribed to cDNA with reverse transcriptase reagents (HaiGene Corp.) according to the manufacturer's protocol. The polymerase chain reaction (PCR) primers of IGF-IR and GAPDH, used as an internal control, were designed using the Primer Expression software 5.0 (Invitrogen Corp.). The sequences used were IGF-IR primer, sense, 5′-TCTGGCTTGATTGGTCTGGC-3′; antisense, 5′-AAC CAT TGG CTG TGC AGT CA-3′; GAPDH primer, sense, 5′-CTG CAC CAC CAA CTG CTT AG-3′; antisense, 5′-TTC TGG GTG GCA GTG ATG-3′. Quantitative real-time PCR was performed with iQ SYBR-Green supermix (Tiangen Corp.). PCR thermal cycling conditions included initial incubation at 95°C for 10 minutes, denaturing at 95°C for 10 seconds, 40 cycles at 60°C for 40 seconds, 95°C for 15 seconds, and 60°C for 1 minute. A melting curve was generated after each PCR reaction to determine the specificity of PCR products. Each sample was tested in triplicate for each gene, and the mean value of the three reactions was used. Real-time PCR results were recorded as Ct values (threshold cycle) and were calculated as ΔCt, which was the difference in Ct values between the target and GAPDH genes. In data analysis, ΔCt was converted to an expression index, using the following formula: 1000 × 2(−ΔCt).
Proliferation assays
Cell proliferation was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay in accordance with the manufacturer's instructions. OVCAR3 cells (5 × 103 cells/well) were seeded in 96-well microplates. The next day, the cells were treated with siRNA. MTT was added (20 μL/well of 5 g/L solution in PBS) after culture for 24, 48, and 72 hours. When incubated at 37°C for 4 hours, the reaction was stopped by addition of 100 μL dimethylsufoxide (Sigma). The dark-blue crystals of MTT–formazan were dissolved by shaking the plates at room temperature for 15 minutes and the absorbance of each well was measured on a Bio-Rad Microplate Reader (Bio-Rad) at a wavelength of 490 nm. Growth inhibition (%) was calculated using the following formula: [1 − (A/B)] × 100, where A is the absorbance of treated cells and B is the absorbance of untreated control cells. All samples were assayed repeatedly in six wells.
Nude mouse xenograft model
Nude mice (female, 4–5 weeks of age) were obtained from Shanghai Laboratory Animal Co. OVCAR3 cells suspended in serum-free DMEM were subcutaneously inoculated (2 × 106/0.2 mL) into the backs of the mice. Tumors were staged for 2 weeks, at which time their volumes were ∼200 mm3. At this time, intratumoral injections were performed with pSih IGF-IR-1 or negative control shRNA (plasmid 0.125 μg/mm3 of tumor volume) or PBS every week. Electroporation method was applied immediately after each injection, the electric voltage of electroporation apparatus was set at 150 V, and the time of impulse was 50 ms.
Tumor volume was determined weekly by external measurements with a caliper and calculated using the following formula: volume = W 2 × L/2, where W is short diameter and L is long diameter. Data are presented as mean ± standard deviation (SD). Animal experiments in the present study were performed in compliance with the guidelines of the Institute for Laboratory Animal Research, Harbin Medical University.
Apoptosis staining
The mice were sacrificed at 35 days postinoculation. The tumors were removed, fixed in 4% polyformaldehyde, paraffin embedded, and sectioned. Apoptotic cells in tumor tissues were detected by TUNEL method using In situ Cell Death Detection Kit (Roche Corporation) according to the manufacturer's protocol. The fractions of apoptotic cells in five random fields from each tumor section were counted, scoring 100 cells in each field, and expressed as an apoptotic fraction (%). Values presented are means (±SD).
Immunohistochemistry
Formalin-fixed, paraffin-embedded blocks from the mouse model were collected in this study. For immunohistochemistry, tissue sections (5 mm) were deparaffinized in xylol and rehydrated in graded alcohol. Endogenous peroxidase activity was blocked with 3% H2O2 for 30 minutes at room temperature. Antigen retrieval was performed by microwave heating the sections in 10 mM sodium citrate buffer (pH 6.0) for 10 minutes at 95°C. Following quenching of endogenous peroxidase activity and blocking of nonspecific binding, slides were incubated with anti-CD31 or anti-Ki-67 antibody (Santa Cruz Biotechnology) at 4°C overnight at a 1:200 dilution. Immunoreactivity was detected using the ABC Peroxidase Kits (Zhongsan Jinqiao Biotechnology Corp.) and 3,3′-diaminobenzidine tetrahydrochloride as the chromogen according to the manufacturer's instructions. Negative controls had all reagents included except the primary antibody. The fraction of Ki-67–positive cells was determined by counting 100 cells in five random fields from each section. Angiogenesis was quantified by counting the number of CD34-positive structures in five randomly selected sections from each tumor section. Mean values (±SD) are presented.
Statistical analysis
All the quantitative data were presented as mean ± SD. The statistical significance of the differences was determined using Student's two-tailed t-test for two groups and one-way ANOVA for multiple groups. All the data were analyzed using SPSS software (version 13.0), and statistical significance was set at p < 0.05.
Results
Effects of IGF-IR siRNA on the expression of IGF-IR from OVCAR3 cells
To inhibit IGF-IR gene expression with siRNA, three plasmids expressing siRNA for IGF-IR were constructed using the pGenesil-1.1 vector. The pGenesil-1.1 vector encodes enhanced green fluorescence protein, so that if the IGF-IR siRNA–expressing plasmids are successfully transfected into the OVCAR3 cells, they can be detected by fluorescence microscopy. As shown in Figure 1, the transfections were successful. IGF-IR protein levels in OVCAR3 cells were quantified by western blot analysis at 48 hours following siRNA exposure. IGF-IR was significantly suppressed to 53.58% (p < 0.001), 88.17% (p < 0.001), and 64.73% (p < 0.001) in cells transfected with pSihIGF-IR-1, pSihIGF-IR-2, and pSihIGF-IR-3, respectively (Fig. 2A), whereas no inhibitory effect was observed in negative control cells (2.04%, p > 0.05).

Transfection of IGF-IR siRNA into cells. The pGenesil-1.1 vector was used to construct plasmids expressing IGF-IR siRNA and GFP as a fluorescence probe. Successfully transfected OVCAR3 cells were easily identified by fluorescence microscopy. IGF-IR, insulin-like growth factor-I receptor; siRNA, small interfering RNA.

Effects of IGF-IR siRNA on IGF-IR expression in transfected cells.
Suppression of IGF-IR expression by the three IGF-IR siRNAs, but not negative control siRNA, was confirmed at the transcript level using real-time PCR at the same time point (Fig. 2B). The expression index of IGF-IR mRNA in groups transfected with siRNAs were 9.500 ± 0.651, 28.260 ± 1.228, and 18.001 ± 0.406, respectively. These results indicated that IGF-IR–specific siRNA can efficiently block expression of IGF-IR at both protein and mRNA levels in human ovarian cancer OVCAR3 cells, and pSih IGF-IR-1 proved to be the most effective at inhibiting IGF-IR expression.
Effects of IGF-IR siRNA on tumor cell proliferation
As shown in Figure 3, compared with untransfected OVCAR3 cells, cell proliferation in R1 group was significantly inhibited to 69.09% at 24 hours (p < 0.001). Cell proliferation in groups R1, R2, and R3 was significantly inhibited to 47.56%, 82.93%, and 73.93% at 48 hours (p < 0.001) and to 50.19%, 86.57%, and 79.21% at 72 hours (p < 0.01), respectively. There was no significant difference between the negative control cells (HK) and untransfected OVCAR3 cells over the entire experimental period (p > 0.05).
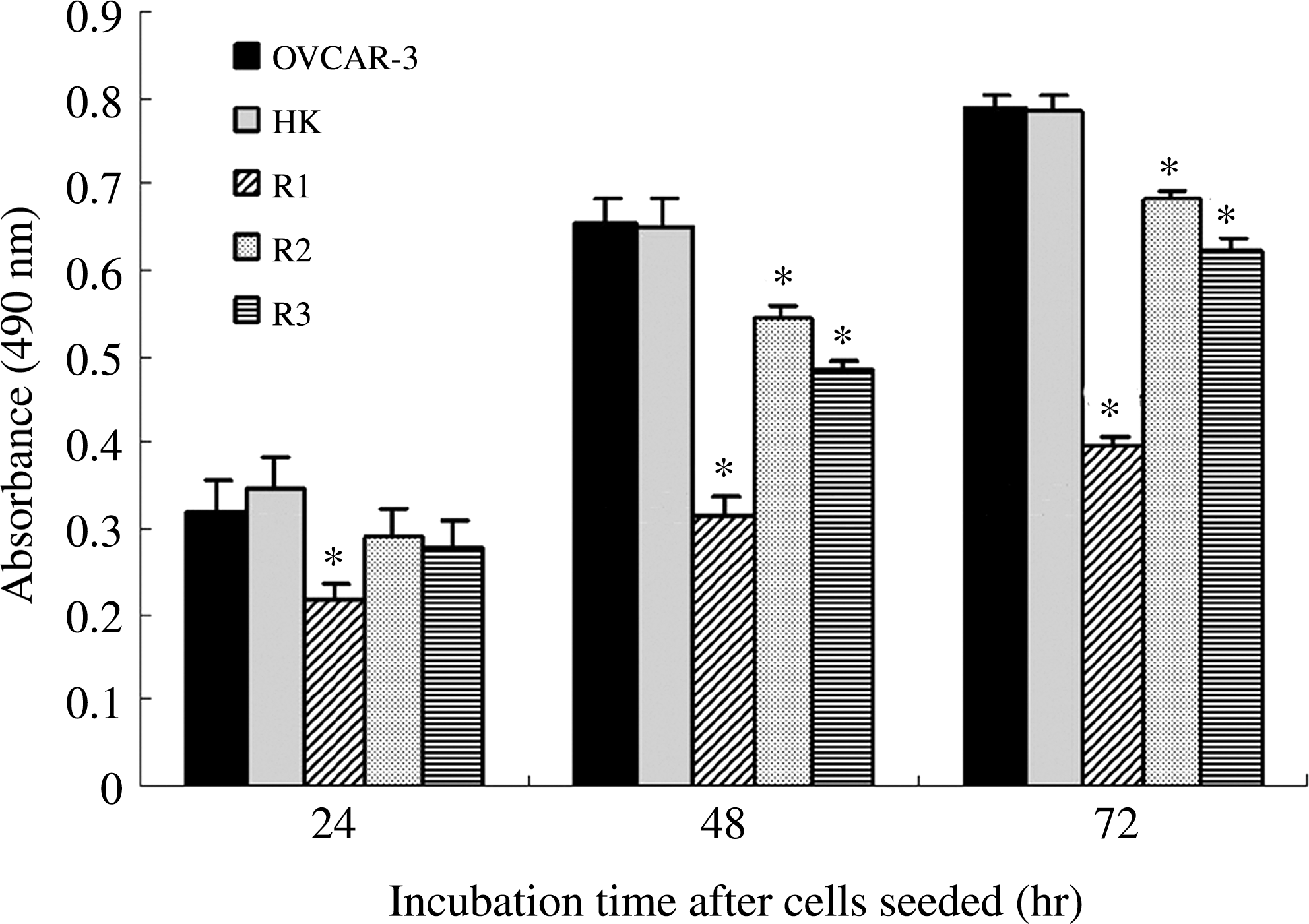
Decrease in the proliferation potential of cells transfected with IGF-IR–targeting siRNA. Cells (OVCAR3, HK, R1, R2, and R3) seeded into 96-well microplates were cultured for 24, 48, and 72 hours and cell numbers were determined by absorbance, as described in the Materials and Methods section. *p < 0.001 versus OVCAR3 cells.
Treatment of the established OVCAR3 xenograft by IGF-IR siRNA
OVCAR3 cells (2 × 106) were injected s.c. into the flank of nude mice. By 2 weeks, visible tumors had developed at the injection sites (mean tumor volume, 243.8 mm3; n = 18). To determine the therapeutic effectiveness of IGF-IR siRNA, intratumoral treatment with pSih IGF-IR-1 was started and repeated every week for 3 weeks. For control groups, PBS and pHK were injected at the same time. A continuous tumor growth was evidenced in mice injected with PBS and pHK, with no difference in tumor sizes between the two groups (p < 0.05). The tumor growth in mice injected with pSih IGF-IR-1 was significantly inhibited when compared with control groups (Fig. 4).
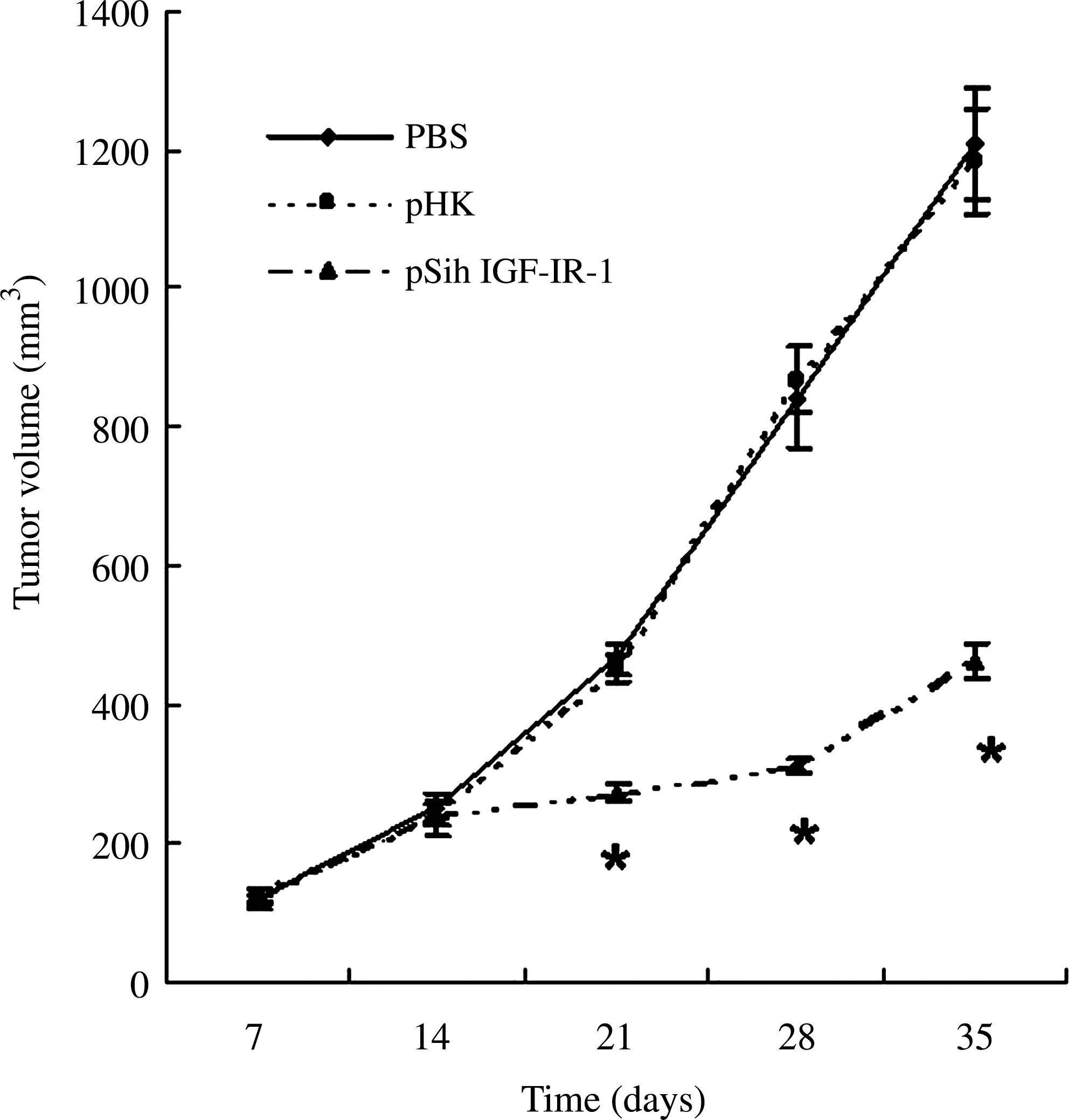
Antitumor effect of IGF-IR siRNA in the OVCAR3 xenograft. Tumor growth curve showed a growth tendency in mice injected with PBS and pHK, whereas the tumor growth in mice injected with pSih IGF-IR-1 was significantly inhibited. A total of 2 × 106 OVCAR3 cancer cells were subcutaneously injected into the backs of the mice and staged for 2 weeks. Intratumoral injections were performed with IGF-IR–specific siRNA–expressing plasmids or negative control siRNA-expressing plasmids or PBS every week. Tumor sizes were measured and calculated as described in the Materials and Methods section. Data are shown as mean ± standard deviation of 6 animals from each group. *p < 0.05. PBS, phosphate-buffered saline.
Tumor cell proliferation and apoptosis in vivo
Tumor apoptosis, determined by TUNEL staining, was increased 2.8-fold in mice treated with IGF-IR siRNA, relative to pHK-treated mice (Fig. 5A). Apoptotic indices of tumors derived from pHK-treated mice did not differ significantly from those of tumors derived from PBS-treated mice. Cellular proliferation, quantified by Ki-67 immunohistochemistry, was reduced by 69% in the tumors from IGF-IR siRNA–treated mice, relative to their control siRNA-treated counterparts (Fig. 5B). Cellular proliferation indices of tumors derived from pHK-treated mice did not differ significantly from those of tumors derived from PBS-treated mice.
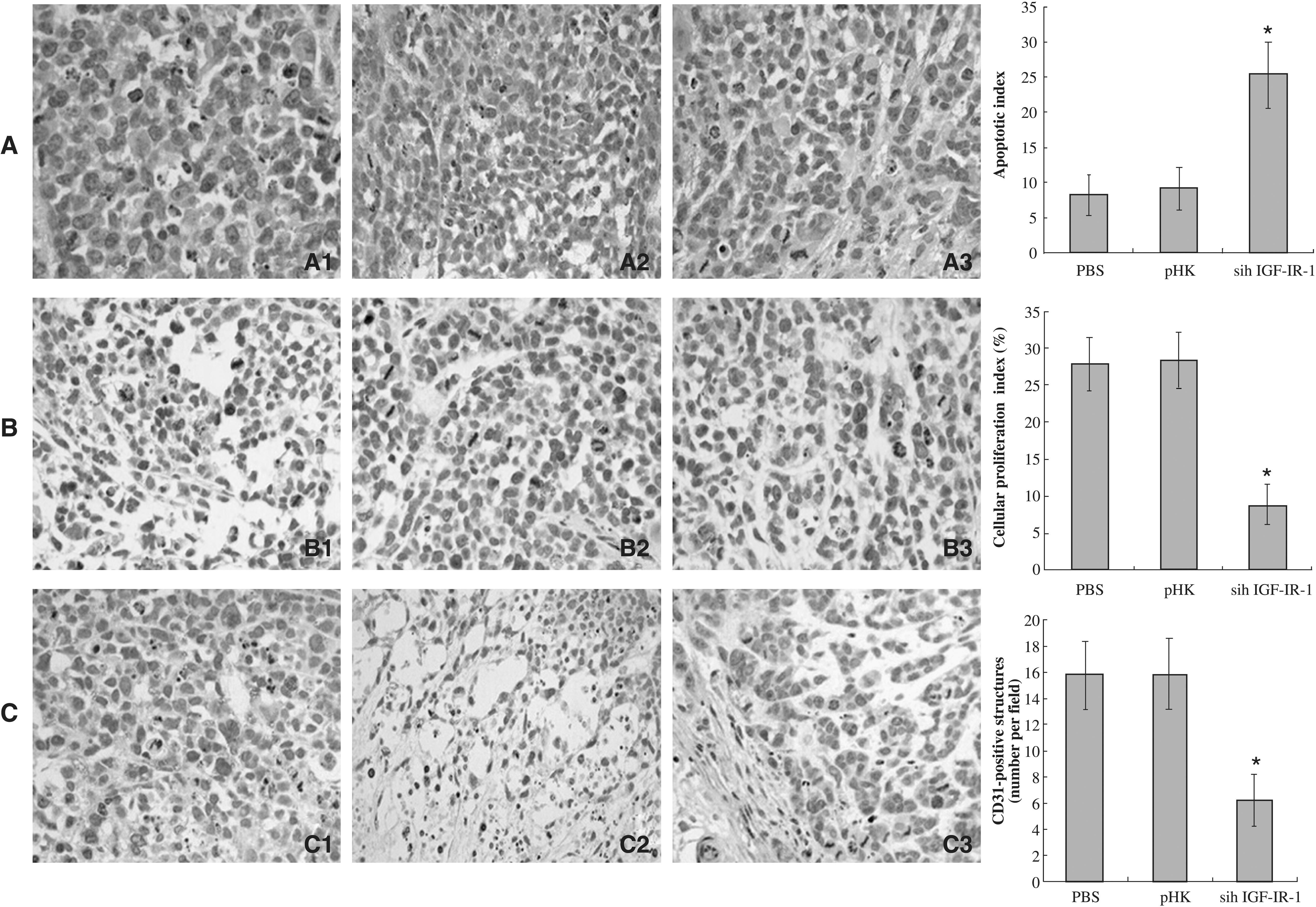
Effect of IGF-IR siRNA on tumor angiogenesis in vivo
Tumor angiogenesis was quantified by immunohistochemical staining with an endothelial cell–specific marker, CD31 (Fig. 5C). Quantification of CD31 staining revealed a significantly decreased microvessel density in tumors transfected with pSih IGF-IR-1 (6.23 ± 1.99) compared with those treated with PBS (15.77 ± 2.58) or pHK (15.8 ± 2.71), respectively. Angiogenesis in tumors from pHK-treated mice did not differ significantly from those observed in tumors from PBS-treated mice.
Discussion
IGF-IR is one of the most tumor-specific genes in the human genome, which is present on all ovarian cancer cells. 10 Its signaling leads to suppression of apoptosis and stimulation of proliferation, and it constitutes an important cell survival pathway. 3,4 Yee et al. 19 identified IGF-IR mRNA in 10 of 10 human ovarian cell lines and in 7 of 7 surgical specimens by RNase protection assay. Beck et al. 9 in 1994 demonstrated IGF-I receptor using an RIA in all primary and metastatic ovarian cancer surgical specimens examined. Experimental studies have shown that malignant transformation of ovarian epithelial cells (the cells from which ovarian cancer is believed to originate) can be induced by overexpression of IGF-IR. 11 It has been reported that CAOV-3 and OVCAR-3 cells produce endogenous IGF-I and grow autonomously in serumless media. The treatment with IGF-IR mRNA antisense oligodeoxynucleotides markedly inhibits the proliferation of these cells both in serumless media and in the presence of IGF-I. When compared with the nontransfected cells, the transfected clone demonstrated a great deal of apoptotic cells and increased expression of Fas-R, a cell membrane protein implicated in the apoptosis signaling pathway. 20 These data indicate that IGF-IR mediates the autocrine proliferation of human ovarian carcinoma cell lines. Inhibition of IGF-IR would result in selective apoptosis and growth inhibition of tumor cells. 3,20,21
In recent years, IGF-IR, as an anticancer target, has been pursued actively because of its importance in tumor cell growth and transformation. 3 Downregulation of IGF-IR function by molecular target therapy has been applied in the treatment of ovarian cancer. Hongo et al. evaluated the antitumor effects of a soluble form dominant negative of the IGF-IR designated as 486/STOP in CaOV-3 human ovarian cancer cells by establishing stable transformants overexpressing 486/STOP and by administration of 486/STOP recombinant protein. Because 486/STOP cells result in massive apoptosis in vivo within 48 hours, usage of a recombinant protein has a great advantage of using its unique bystander effect in vivo for clinical application. 14 Gotlieb et al. investigated the antineoplastic activity of NVP-AEW541, a small-molecular-weight inhibitor of the IGF-IR kinase, and presented that IGF-IR is a potential new molecular target in ovarian cancer. 15 However, few reports have been found about the inhibitory effects of siRNA against IGF-IR in ovarian cancer in vitro and in vivo. 15
In gene function studies, the specific knock-down of target genes without affecting other genes is critically important. RNAi is a specific gene-silencing technology. This technology was used to construct the IGF-IR siRNA expression plasmids for transfection into the ovarian cancer cell line OVCAR3. Three plasmids expressing siRNA for IGF-IR were constructed. The pSih IGF-IR-1, pSih IGF-IR-2, and pSih IGF-IR-3 reduced expression of IGF-IR significantly at both protein and mRNA levels. The inhibitory effect of pSih IGF-IR-1 was the greatest. The present study postulates that positional effects and a variance in the secondary structure of the nucleotide sequence at different sites may play a role in the different inhibitory effects of the siRNA targeting the same gene.
Unlike the antisense technique, which has no amplification effect and limited inhibition effect range of 10%–30%, 22 RNAi lasts for multiple cell cycles before restoration of gene expression. In the present study, it has been found that RNAi against IGF-IR successfully inhibited the expression of IGF-IR in human OVCAR3 cells, leading to a potent suppression of tumor cell proliferation in vitro. The suppressive effect of siRNA to IGF-IR can reach up to nearly 50%. This is consistent with recent studies that showed that the introduction of a 21-nt double-stranded RNA into cancer cells strongly suppresses the expression of specific mRNAs. 17,23 In the nude mouse xenograft model, intratumorally administered IGF-IR–specific siRNA suppressed tumor growth and cellular proliferation, as well as promoted tumor cellular apoptosis and inhibited angiogenesis. The effects on malignant cellular behavior and tumor progression were marked, suggesting the importance of IGF-IR expression as a determinant of ovarian cancer tumorigenicity in the nude mouse xenograft model. The effects of IGF-IR–specific siRNA on tumor angiogenesis is intriguing and may underlie an as yet unrecognized function of IGF-IR. There are clearly limitations to the nude mouse as a model for human cancer, but the results support further investigation of siRNA as a therapeutic strategy in human patients.
Conclusions
The present study demonstrated that plasmid-mediated RNAi of IGF-IR effectively inhibits the growth of ovarian cancer in vitro and in vivo. The RNAi approach can be used as an effective therapeutic strategy for ovarian cancer. The present study provides a basis for future studies of this approach in additional animal models and in human clinical trials.
Footnotes
Acknowledgments
This work was partially supported by the scientific and technological project of Heilongjiang Province, China (No. GB07C32304).
Disclosure Statement
No competing financial interests exist.