
Abstract
Human UDP-glucuronosyltransferases (UGTs) are a family of membrane-bound enzymes of the endoplasmic reticulum. They catalyze the glucuronidation of various endogenous and exogenous compounds, converting them into more polar glucuronides. In this study, uracil glucuronide was enzymatically synthesized using a UGT-rich microsome preparate, which was separated from Hutu-80 cells. Two different glucuronide derivatives were obtained, with a total reaction yield of 22.95% ± 2.4% (n = 4). The glucuronide ligands were defined as uracil-n-glucuronide (UNG) and uracil-o-glucuronide (UOG). These were then analyzed by high-performance liquid chromatography–mass spectrometry and labeled with I-125 and I-131, separately. The radiolabeled 125/131I-UNG and 125/131I-UOG presented good incorporation ratios for Hutu-80, Caco-2, Detroit 562, and ACBRI 519 cells. The incorporation ratios of 125/131I-UOG were higher than those of 125/131I-UNG and of other labeled components for all cell types, and were also statistically significant compared to the values of 125/131I-UNG for primary human intestinal epithelial cells (ACBRI 519) and human intestinal adenocarcinoma cells. Cell incorporation rates of n-glucuronides and o-glucuronides were higher compared to uracil, with o-glucuronides being more selective. The results suggest that both I-125- and I-131-labeled glucuronides can be used in imaging and therapy, and further research should be done in preclinical stages.
Introduction
Lack of selectivity of drugs is a major limitation in cancer chemotherapy. Most drugs are not selective enough to be used in optimal doses without severe drawbacks. A solution is to target the active drugs to the cancer cells. 1 Selectivity at the tumor site in cancer therapy could be conveyed by conjugates of monoclonal antibodies and enzymes, which liberate the toxin from the prodrug. 2 These antibodies have been used as carriers of cytotoxic agents, such as conventional cytotoxic drugs, radionuclides, or toxins. Monoclonal antibodies can also be used for tumor-selective delivery of an enzyme that converts a relatively nontoxic prodrug into its toxic form. The selective activation of the prodrug at the site of the tumor enhances drug concentration in the tumor, resulting in a more successful antitumor effect and a reduction in systemic toxicity. 3
Certain enzymes in the human tissues and body fluids may play a role in the detoxification process and may influence the composition and availability of steroid hormones, toxins, or carcinogens. 4 One of the most important detoxification processes occurs through glucuronidation conjugation. Glucuronidation is one of the main phase-2 metabolic pathways by which an organism transforms a drug or a xenobiotic into a more water-soluble substance, allowing its detoxification and subsequent excretion. 5 Glucuronides of drugs often accumulate during long-term therapy. The enzyme β-glucuronidase catalyzes the hydrolysis of glucuronides and is also helpful in tumor-specific bioactivation of glucuronide prodrugs of anticancer agents. 6 Glucuronidation is an SN2 reaction where the configuration of the glucuronic acid changes from α- to β-anomer. 7 o- and n-glucuronides are the most common and C- and S-glucuronides are the most rare drug glucuronides in humans. 7,8 Glucuronide prodrugs have potent antitumor activity in antibody-directed enzyme prodrug therapy, where specific antibodies are used to deliver β-glucuronidase to cancer cells. β-Glucuronidase is also used directly in prodrug monotherapy, which relies on the presence of elevated levels of this enzyme in the tumor interstitial space. 9 Glucuronides are one of the major metabolites produced in humans. These and other metabolites are also important in studies of the metabolism of new drugs, because the metabolites possibly have toxic or other undesirable effects completely different from those of the original drug. 10
The aim of this study was to synthesize glucuronide derivatives of uracil by using enzyme fractions separated from human cancer cells, to label these derivatives with radioiodine, and to develop new agents for tumor imaging and therapy by examining the efficiency of these radiolabeled compounds on cancer cells.
Materials and Methods
Materials
Na131I was obtained from the Nuclear Medicine Department of Ege University. Na125I was obtained from the Institute of Isotopes Co. Ltd. Uracil was purchased from MP Biomedicals. Hutu-80, Caco-2, and Detroit 562 were obtained from the American Type Culture Collection. Primary human intestinal epithelial cell (ACBRI 519) was obtained from the Applied Cell Biology Research Institute. All other chemicals were supplied by Merck Chemical Co. and Aldrich Chemical Co.
Thin-layer radiochromatography (TLRC) was performed using a Bioscan AR-2000 Imaging Scanner. Liquid chromatography–mass spectrometry (LC/MS/MS) chromatograms were taken using a TandemGold Triple Quadrupole LC/MS/MS instrument.
Enzymatic synthesis of uracil glucuronide
UDP-glucuronyl transferase (UDPGT)-rich microsomal fractions separated from Hutu-80 (human duodenum adenocarcinoma cell line) were used for glucuronidation reaction of uracil.
Separation of microsomal fractions from Hutu-80 cells
Hutu-80 cells were grown in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 1.5 g/L sodium bicarbonate, 0.1 mM nonessential amino acid, and 1 mM sodium pyruvate in a humidified 5% CO2 incubator at 37°C. The cells were maintained in exponential growth by subculturing them with trypsin–ethylenediaminetetraacetic acid (EDTA) (0.025% by w/v in Hanks' balanced salt solution). The cells were then pelleted and resuspended in cell medium. Microsomal fractions were separated from the cells as described by Zihnioglu. * Grown cells were treated with trypsin-EDTA, resuspended in cell medium, and placed in ice-cold 250 mM sucrose/5 mM HEPES 99% [4-(2-hydroxyethyl)-1-piperazineethane-sulfonic acid, sodium salt], 0–4°C. The solution was then homogenized after adding 250 mM sucrose/5 mM HEPES (pH 7.4) for 30 minutes.
Purification of UDPGT from microsomal fractions of Hutu-80 cells
The cell homogenate was centrifuged at 12,000 g for 10 minutes. The resulting supernatant was separated and then centrifuged at 105,000 g for 1 hour at +4°C. Microsomal pellets were dissolved in a mixture of 0.2 M potassium phosphate, 2 mM mercaptoethanol, and 0.4% TritonX100 (pH 7) buffer. The suspension was stirred on ice for 30 minutes and centrifuged at 105,000 g for 1 hour at +4°C to remove insoluble material. The resulting supernatant was stored at −80°C.
Determination of protein quantity in microsomal samples
Protein content in the samples was determined with the bicinchoninic acid (BCA) reagent (Pierce). Twenty-five (25) microliters of the assay sample was added to 200 μL of BCA reagent. Standards and samples were measured spectrophotometrically at 562 nm after incubating at 37°C for 30 minutes. Standard curves, using bovine serum albumin (Sigma), were plotted by linear regression analyses. Protein concentration of appropriately diluted samples calculated from the relevant standard curve was ∼153.11 mg/mL.
Glucuronidation reaction
The procedure used for glucuronidation reaction was similar to that reported in other studies. 11,12 One hundred (100) microliters of the microsomal enzyme preparate (153.11 mg protein/mL) was added to 5 mL of 50 mM Tris buffer (pH 8.0) containing 6 mM CaCl2, 10 mM UDPGA, and 1 mM dithiothreitol at 37°C. The mixture was stirred at 37°C in a water bath for 10 minutes. The contents were then sonicated in an ultrasonic bath for 30 seconds to disperse the microsomes and the reactions. This solution was incubated by slow stirring at 37°C for 18 hours after adding 10 mg/mL of uracil in dimethyl sulfoxide. The reaction was terminated after 18 hours by adding 300 μL of acetonitrile, and the precipitated protein was removed by centrifugation at 6000 g for 10 minutes. The supernatant was then analyzed by reversed-phase high-performance liquid chromatography (HPLC) (Shimadzu 10 AVp). Two different glucuronide derivatives were obtained, with a total reaction yield of 22.95% ± 2.4% (n = 4). The glucuronide ligands were defined as uracil-o-glucuronide (UOG) and uracil-n-glucuronide (UNG) by LC/MS/MS.
LC/MS/MS parameters
LC/MS/MS chromatograms were taken using a Tandem Gold Triple Quadrupole LC/MS/MS instrument. The LC/MS/MS system consisted of a Zivak P-100 Dual Pump, a Zivak AS-100 Autosampler module, and a Zivak Tandem Gold Triple Quadrupole Tandem Mass Spectrometer with an ESI ionization module. The parameters were optimized and set as follows: positive electrospray ionization with a spray voltage of 5000 V, drying gas temperature of 350°C, spray chamber temperature of 350°C, drying gas pressure of 35 psi, nebulizing gas pressure of 51 psi, and a detector of 1500 V. HPLC was performed on a 150 × 2 mm Synergy 4u Max-RP 80A column (Phenomenex 00F-4337-B0). This was maintained at 30°C using a mixture of water with 0.1% formic acid and acetonitrile with 0.1% formic acid (60:40) as a mobile phase, which was delivered at 0.25 mL/min.
Labeling of uracil glucuronide derivatives with 125/131I
Uracil, UOG, and UNG were separately labeled with 125I and 131I by the iodogen method. 13,14 For labeling with 125I, 0.5 mg uracil, 0.1 mg UOG, and 0.1 mg UNG were added into the iodogen-coated tubes. About 2–3 mCi of Na125I was then added. The reaction mixture was kept at room temperature without stirring for 15 minutes and then transferred into other iodogen-coated tubes consecutively. For labeling with 131I, the same procedure as described above for 125I was repeated. Quality control procedures were performed in each case.
Quality control procedures
High-performance liquid chromatography
A low-pressure-gradient HPLC system (an LC-10ATvp quaternary pump, an SPD-10A/V UV detector, a syringe injector equipped with a 1-mL loop, and a 7-μm RP-C18 column of 250 × 21 mm ID; Macherey–Nagel) was used for preparative procedures. The elute was collected with an FRC-10A fraction collector (Shimadzu). The flow rate was set at 9.0 mL/min.
For analytical experiments, a 5-μm RP-C18 column (250 × 4.6 mm ID) (Macherey–Nagel) and a syringe injector equipped with a 20-μL loop were used. UV detection was achieved at 254 and 280 nm. The column oven temperature was set at 30°C. A 10 μL sample was applied to the column and eluted with 50% 3 × 10−3 M tetramethyl ammonium hydroxide and 50% 3 × 10−3 M formic acid (pH 8) at 0.4 mL/min.
Thin layer radiochromatography
For thin layer radiochromatography (TLRC) studies, TLC silica sheets (Merck; 20 × 20 cm; #1.05554) were used. Three different baths, consisting of n-butanol/H2O/acetic acid (4:2:1), ethyl alcohol/H2O (3:2), and isopropyl alcohol/n-butanol/0.2 N NH4OH (4:2:1), were used as the mobile phase. Each TLRC sheet was covered by an adhesive band after its development and subsequently cut into 0.5-cm widths. These pieces were counted using a Bioscan AR-2000 Imaging Scanner. The TLRC chromatograms were obtained from these countings. The retention factor (R f ) values and labeling efficiencies were derived from these chromatograms.
Paper electrophoresis
Electrophoresis was performed with a Gelman Electrophoresis Chamber supply using cellulose acetate strips. Cathode and anode poles as well as application points were indicated on cellulose acetate strips, which were then moistened by n-butanol/H2O/acetic acid (4:2:1) solution. The strips were then placed in the electrophoresis chamber after the samples were set on them. Standing time and applied voltage were 120 minutes and 300 V, respectively. The developed strips were dried and counted by a Bioscan AR-2000 Imaging Scanner.
Lipophilicity
Lipophilicity (logP; P being the partition coefficient) was determined for 131I-uracil, 131I-UOG, and 131I-UNG. Two hundred (200) microliters of the radiolabeled compound was added to a premixed suspension of 200 μL of octanol in 200 μL of water. The resulting solution was vortexed at room temperature for 1 hour and then centrifuged for 30 minutes at 2500 rpm. The octanol and water layers were separated from each other, radioactivity of each layer was measured, and the layers were then weighed. P was calculated as the ratio of counts per second (cps)/g of octanol to cps/g of water. The average logP value for five trials was then reported. Calculated values were compared with the theoretical lipophilicity values obtained by the additive consecutive algorithm (Advanced Chemistry Development, Inc., Toronto, Ontario, Canada) computer program.
Serum stability
The stabilities of uracil, UOG, and UNG were investigated under physiological conditions. Blood serum obtained from a healthy volunteer adult was used. One hundred (100) microliters (30 μg) of the labeled sample was added to 300 μL of fresh human serum in triplicate at 37°C throughout 24 hours. During incubation, the samples were analyzed by the TLRC method at 30, 60, and 240 minutes, and at 24 hours.
Cell culture
Hutu-80, Caco-2, Detroit 562, and ACBRI 519 cell lines were used for cell culture studies. Hutu-80 cells were grown in EMEM supplemented with 10% FBS, 2 mM glutamine, 1.5 g/L sodium bicarbonate, 0.1 mM nonessential amino acid, and 1 mM sodium pyruvate. Caco-2 cells were cultured in EMEM supplemented with 20% FBS and the same other reagents as used for Hutu-80. Detroit cells were grown in EMEM supplemented with the reagents used for Hutu-80 cells, along with 0.1% lactalbumin. ACBRI 519 cells were cultured in complete serum-containing medium kits. In all experiments, cells were grown at 37°C in a humidified incubator equilibrated with 5% CO2. The cells were maintained in exponential growth by subculturing with trypsin-EDTA (0.25% by w/v in Hanks' balanced salt solution). Cells were then pelleted and resuspended in cell medium.
To obtain the enzyme UDPGT, microsomal fractions of Hutu-80 cells were separated and synthesis of uracil glucuronide was performed using this enzyme. Therefore, time- and dose-dependent incorporation of UOG, UNG, and uracil were investigated using Hutu-80 cells. Studies were performed six times for each experimental condition.
Time-dependent incorporation of 125I-UOG, 125I-UNG, and 125I-uracil
Thirty (30) micrograms of each sample labeled were combined with 4 μCi/mL of Na125I per well using the iodogen method. The trypan blue exclusion method was used to assess cell viability. Hutu-80 cells were seeded in 24-well plates at ∼1.0 × 105 cells per well and cultured to confluence. Monolayers were washed three times with phosphate-buffered saline (PBS), and I-125-labeled samples (0.5 mL, 30 μg/mL in a culture medium) were then added to the cells. I-125 was also added to the cells to check its incorporation. Additionally, 0.5 mL of labeled samples in a culture medium was used as a control group for radioactivity measurement. Due to investigation time dependent incorporation of 125I-UOG, 125I-UNG, and 125I-uracil, the cells were incubated for 10, 30, 60, and 120 minutes. After incubation, the cells were washed three times with PBS and suspended by treating with 200 μL of RIPA lyse buffer solution. Part of the lysed cell suspension (50 μL) was used for determination of protein quantity by the BCA method, and 100 μL was collected for radioactivity measurement by counting cells in a Packard Tri-corb-1200 liquid scintillation counter.
Dose-dependent incorporation of 125I-UOG, 125I-UNG, and 125I-uracil
To investigate the optimum dose-dependent incorporation of UOG, UNG, and uracil, various concentrations of samples were treated with cells in a culture medium. Thirty (30) micrograms of each sample was labeled with 4 μCi/mL of 125I for the time-dependent incorporation study. Therefore, 1, 3, 10, 30, and 100 μg/mL of samples were labeled, respectively, with 0.1, 0.4, 1.3, 4, and 13.2 μCi/mL of 125I for the use of equal activity per molecule. Optimum incubation time was defined as 30 minutes based on the result of the time-dependent incorporation study. Labeling and cell culture experiments were conducted on Hutu-80 cells according to the procedure explained in the time-dependent incorporation study.
Incorporation of 125I-UOG, 125I-UNG, and 125I-uracil on three different adenocarcinoma cells
Incorporation of uracil glucuronide derivatives on Hutu-80 (human duodenumintestinal adenocarcinoma), Caco-2 (human colon adenocarcinoma), and Detroit 562 (human pharynx adenocarcinoma) cells was performed using the enzyme UDPGT (obtained from Hutu-80 cells) to investigate differences between the incorporation of these various types of adenocarcinoma cell lines. Labeling and cell culture studies were performed at the optimum conditions for incubation time and dose. Cell culture experiments were conducted by following the procedure of the time-dependent incorporation study.
Incorporation of 131I-UOG, 131I-UNG, and 131I-uracil on three different adenocarcinoma cells
To see the decay effect of radioiodine, the experiments described above were repeated on the three different adenocarcinoma cell lines (Hutu-80, Caco-2, and Detroit 562) under the same conditions using I-131 instead of I-125 for labeling the compounds.
Incorporation of I-125-labeled samples on normal cells
At this stage, incorporations of I-125-labeled samples on adenocarcinoma cells and on normal cells were compared. The human duodenum adenocarcinoma cell line (Hutu-80) and the primary human intestinal epithelial cell (ACBRI 519) were used for comparison. Cell lines were cultured and samples labeled with I-125. Cell culture experiments were conducted under optimum conditions according to the method indicated in the time-dependent incorporation study.
Effect of increasing specific activity of 125/131I on incorporation of labeled samples
To investigate the effect of increasing the specific activity of 125/131I, samples were labeled separately with five different specific activities of I-125 and I-131 using the iodogen method and incorporated on Hutu-80 cells. Thirty (30) micrograms of each sample was labeled separately with 1, 3, 10, 30, and 100 μCi/mL of I-125 and I-131 per well in 24-well plates isolated from each other.
Hutu-80 cells were implanted in 24-well plates at ∼1.0 × 105 cells per well and cultured to confluence. Monolayers were washed three times with PBS, and the 125/131I-labeled samples of different specific activities (0.5 mL, 30 μg/mL in culture medium) were then added separately to the cells. Each used activities (1, 3, 10, 30, 60, and 120 minutes) of I-125 or I-131 were also added to the cells. Additionally, 0.5 mL of labeled samples in a culture medium was used for radioactivity measurement as a control group. The cells were incubated for 30 minutes and then washed three times with PBS solution. Subsequently, the cells were suspended by treating with 200 μL of RIPA lyse buffer solution. Fifty (50) microliters of the lysed cell suspension was used for the determination of protein quantity, and 100 μL of the cell suspension was used for radioactivity measurement.
Statistical analyses
Statistical significance was assessed with one-way analysis of variance and linear regression by the GraphPad program. p-Values <0.05 were considered statistically significant.
Results
HPLC chromatograms of uracil and its glucuronide derivatives showed two glucuronide peaks, as seen in Figure 1. The total reaction yield was 22.95% ± 2.4% (n = 4). The HPLC chromatograms were compared with theoretical lipophilicities (Figure 2). The first and the second peaks were defined, respectively, as UOG and UNG (Figures 3 and 4). The last peak was identified as uracil. These three peaks were separated under preparative HPLC conditions and, as expected from theoretical lipophilicities, separation occurred at the reversed phase of HPLC.

High-performance liquid chromatography chromatogram of uracil and uracil glucuronide derivatives. UNG, uracil-n-glucuronide; UOG, uracil-o-glucuronide.

Theoretical high-performance liquid chromatography chromatogram of uracil-o-glucuronide, uracil-n-glucuronide, and uracil according to theoretical lipophilicity.
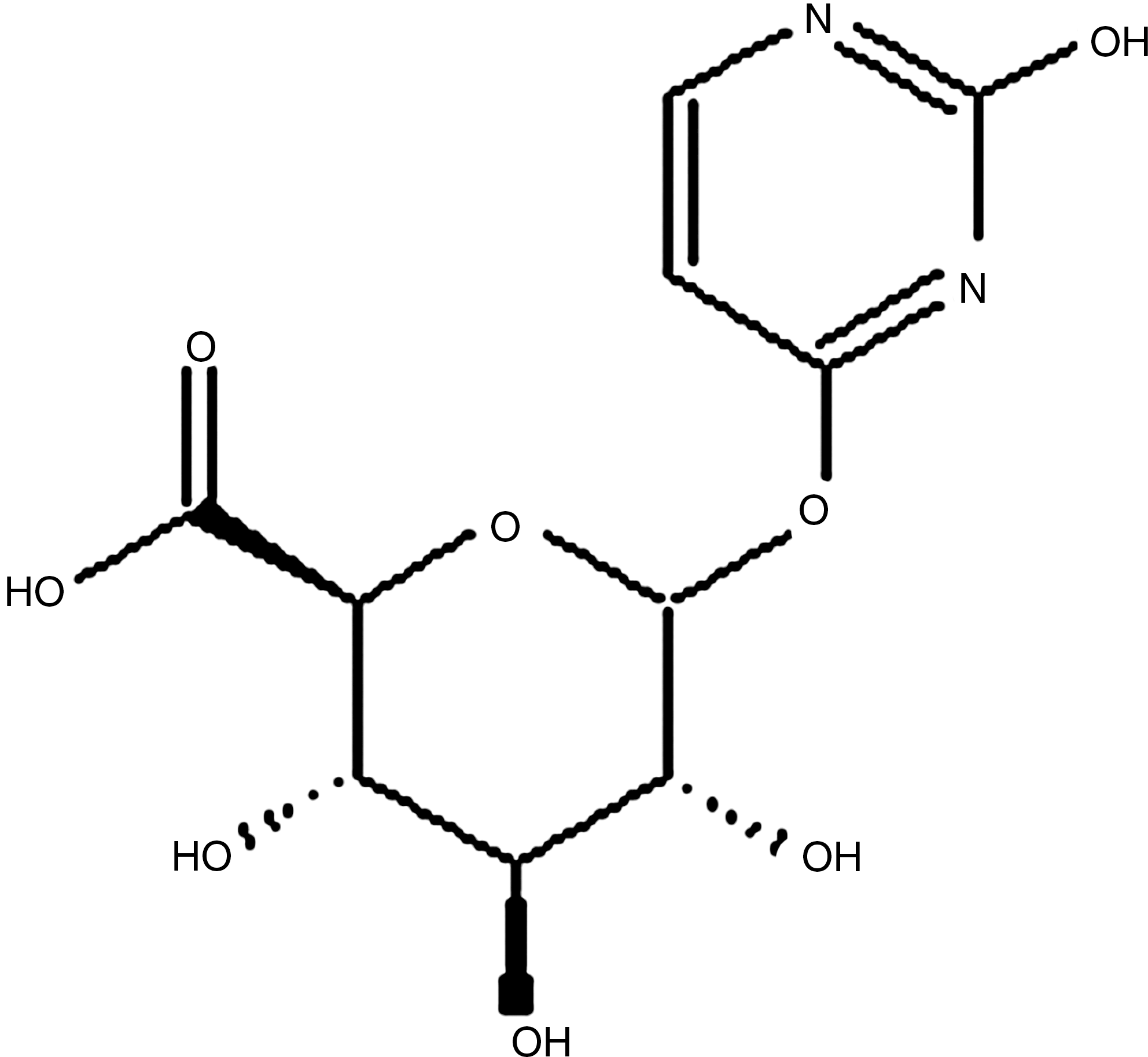
Uracil-o-glucuronide.
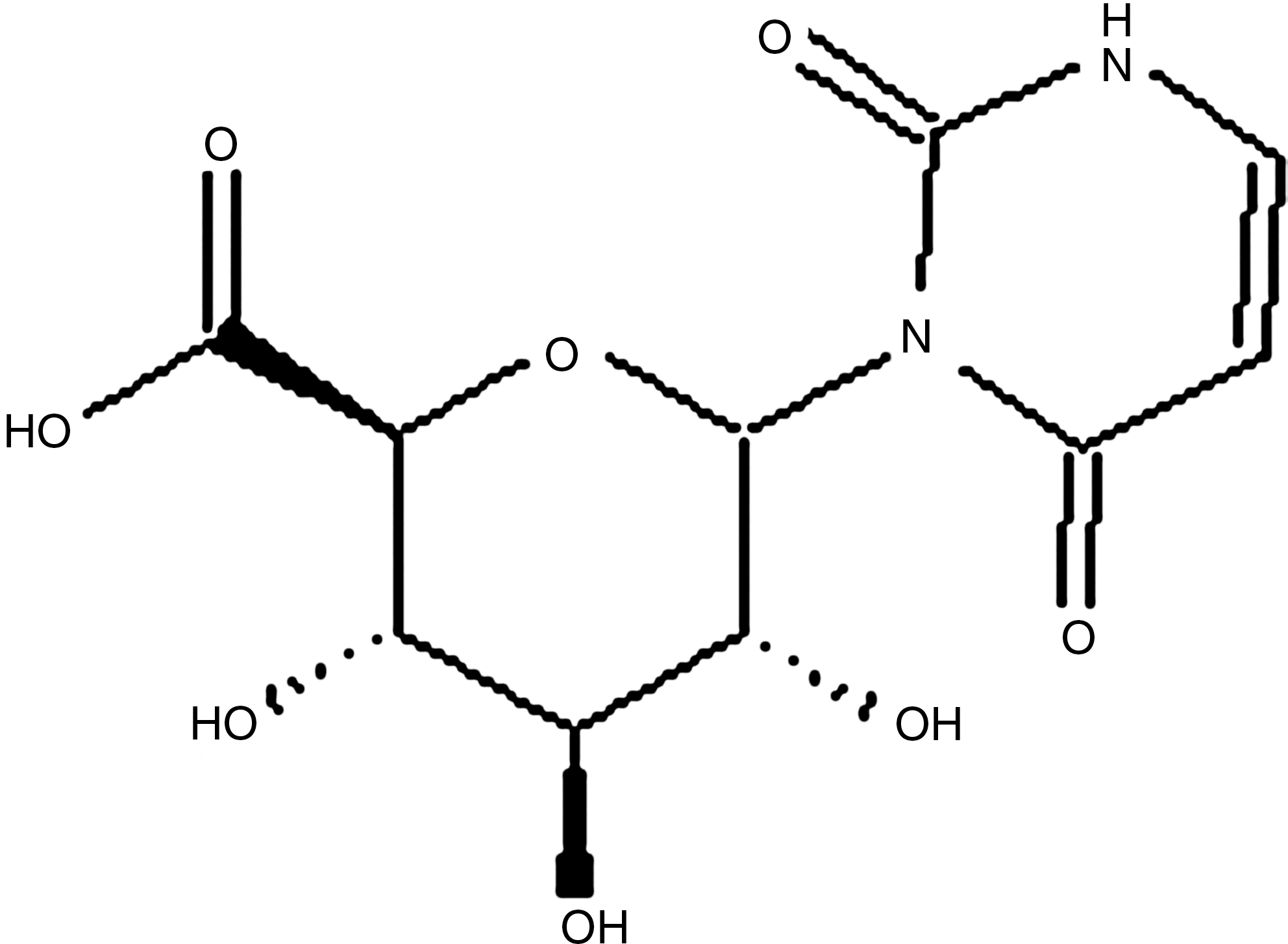
Uracil-n-glucuronide.
The separated peaks were lyophilized and then analyzed by LC/MS/MS (ZIVAK-TANDEM GOLD LC-MS/MS). Results confirmed the expected structures. As shown in Table 1, the molecular fragments at m/z 112.08 belong to uracil; those at 148.15, 172.13, and 228.20 belong to UOG; those at 142.11, 150.13, and 228.20 belong to UNG. Figures 5 and 6 show positive ESI MS/MS spectra of UOG and UNG, respectively.
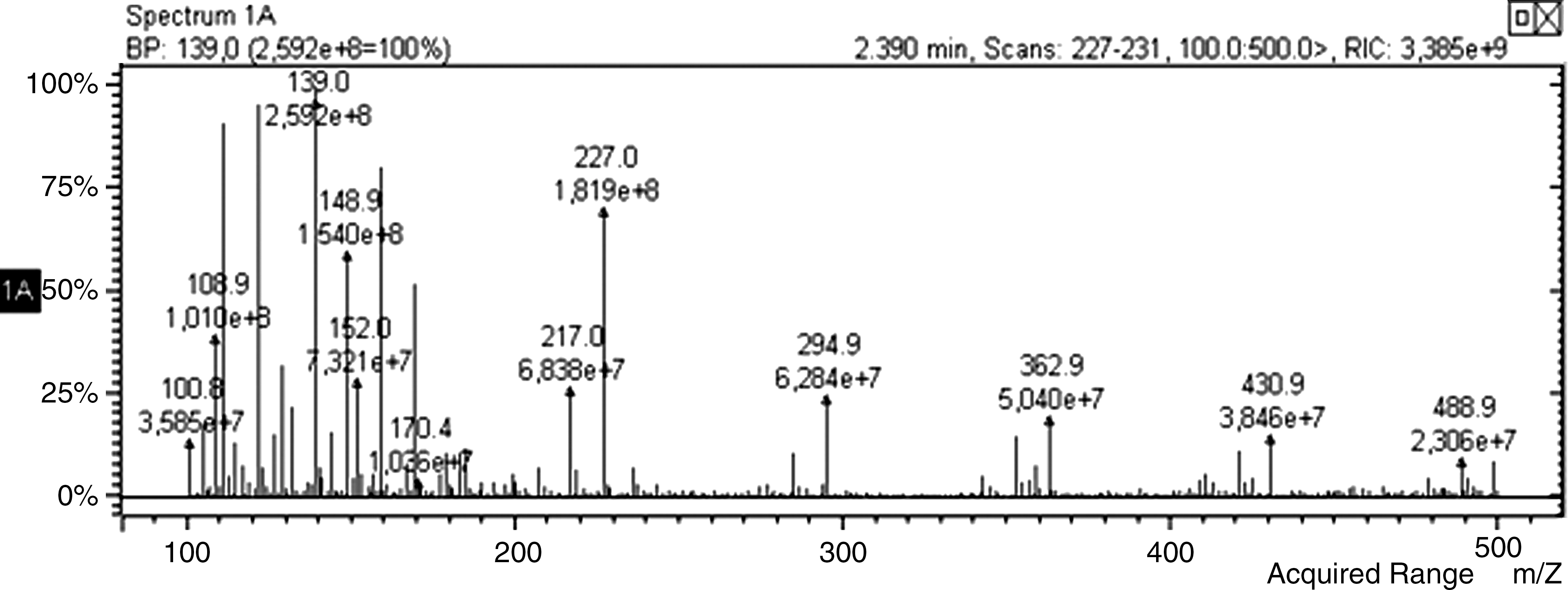
Positive ESI MS/MS of uracil-o-glucuronide.
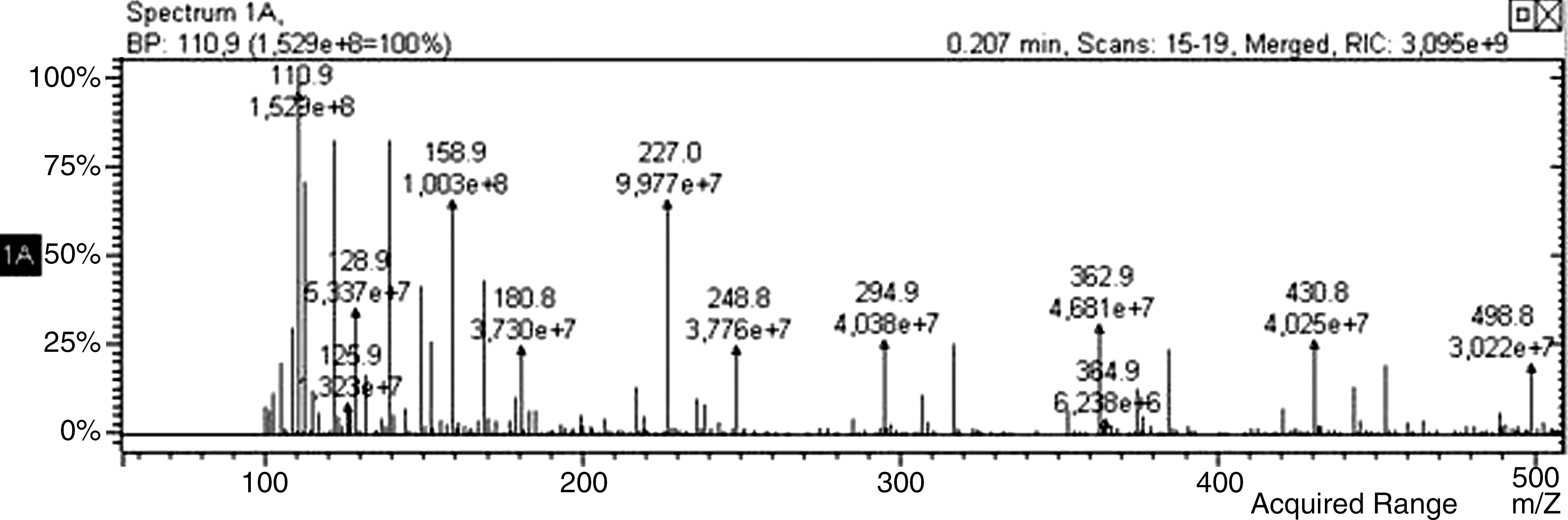
Positive ESI MS/MS of uracil-n-glucuronide.
Rr values of I-131 labeled compounds were given in Table 2. The labeling yields of 131I-UOG and 131I-UNG were 96.91% ± 0.9% (n = 10) and 96.01% ± 1.7% (n = 10), respectively, as shown in Table 3. R f values of I-125 labeled uracil and uracil glucuronide deriveratives are different from each other, as given in Table 4. Labeling yields of I-125-labeled compounds were also above 90% (n = 10), as shown in Table 5. According to electrophoresis results, as seen in Table 6, 125/131I-UOG and 125/131I-UNG have neutral structures.
Developing medium: n-butanol/H2O/acetic acid (4:2:1).
Developing medium: ethyl alcohol/H2O (3:2).
Developing medium: isopropyl alcohol/n-butanol/0.2 N NH4OH (4:2:1).
TLRC, thin-layer radiochromatography; UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
Developing medium: n-butanol/H2O/acetic acid (4:2:1).
TLRC, thin-layer radiochromatography; UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide; TLRC, thin-layer radiochromatography.
The lipophilicities for 131I-UOG, 131I-UNG, and 131I-uracil were − 5.05 ± 0.91, −2.13 ± 0.72, and +0.03 ± 0.02 (n = 5), respectively, while the corresponding theoretical values according to the ACD/logP algorithm program were −7.40, −4.70, and +0.04. In vitro stability of the radiolabeled uracil glucuronide derivatives was quite high. Labeling yields of 131I-UOG and 131I-UNG were 91.28% ± 3.2% and 90.28% ± 2.3% after 24 hours, respectively. Hence, the period of stability of these radioiodinated derivatives was sufficient for cell culture study.
The time-dependent incorporation values of the I-125-labeled compounds are shown in Figure 7. The incorporation of 125I-UOG significantly differed from that of other compounds after 30 minutes (p < 0.05). Incorporation of 125I-UOG and 125I-UNG were higher than those of 125I-uracil and I-125 at all time periods. Although 125I-UOG incorporated more than 125I-UNG, the difference was not statistically significant except after 30 minutes. Optimum incorporation time for all labeled compounds was determined as 30 minutes, which was significantly different between all compounds.
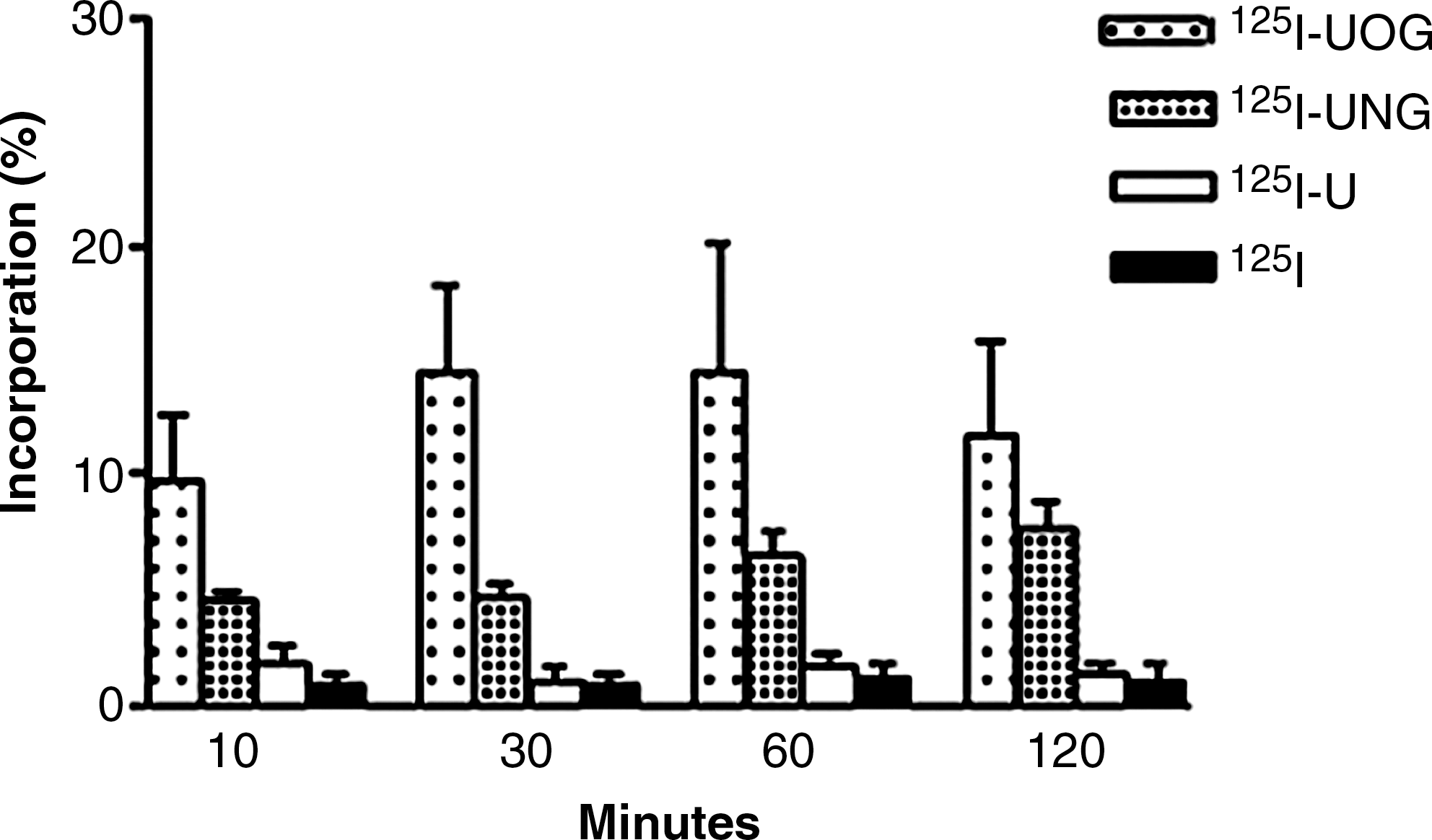
Time-dependent incorporation values of I-125-labeled compounds. U, uracil; UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
The dose-dependent incorporation values of the I-125-labeled compounds are shown in Figure 8. The incorporation of 125I-UOG at 30 μg/mL was significantly different (p < 0.05) from that at other doses. There was a statistically significant difference (p < 0.05) between the incorporation of 1, 10, and 30 μg/mL of 125I-UNG. However, there was no significant difference between the incorporation of I-125 at different doses. Incorporations of 125I-UOG were higher than those of 125I-UNG, 125I-uracil, and I-125 at all doses. 125I-UOG and 125I-UNG had higher incorporation values than 125I-uracil and I-125. Optimum incorporation time and dose were determined as 30 minutes and 30 μg/mL, respectively, for other cell culture experiments.
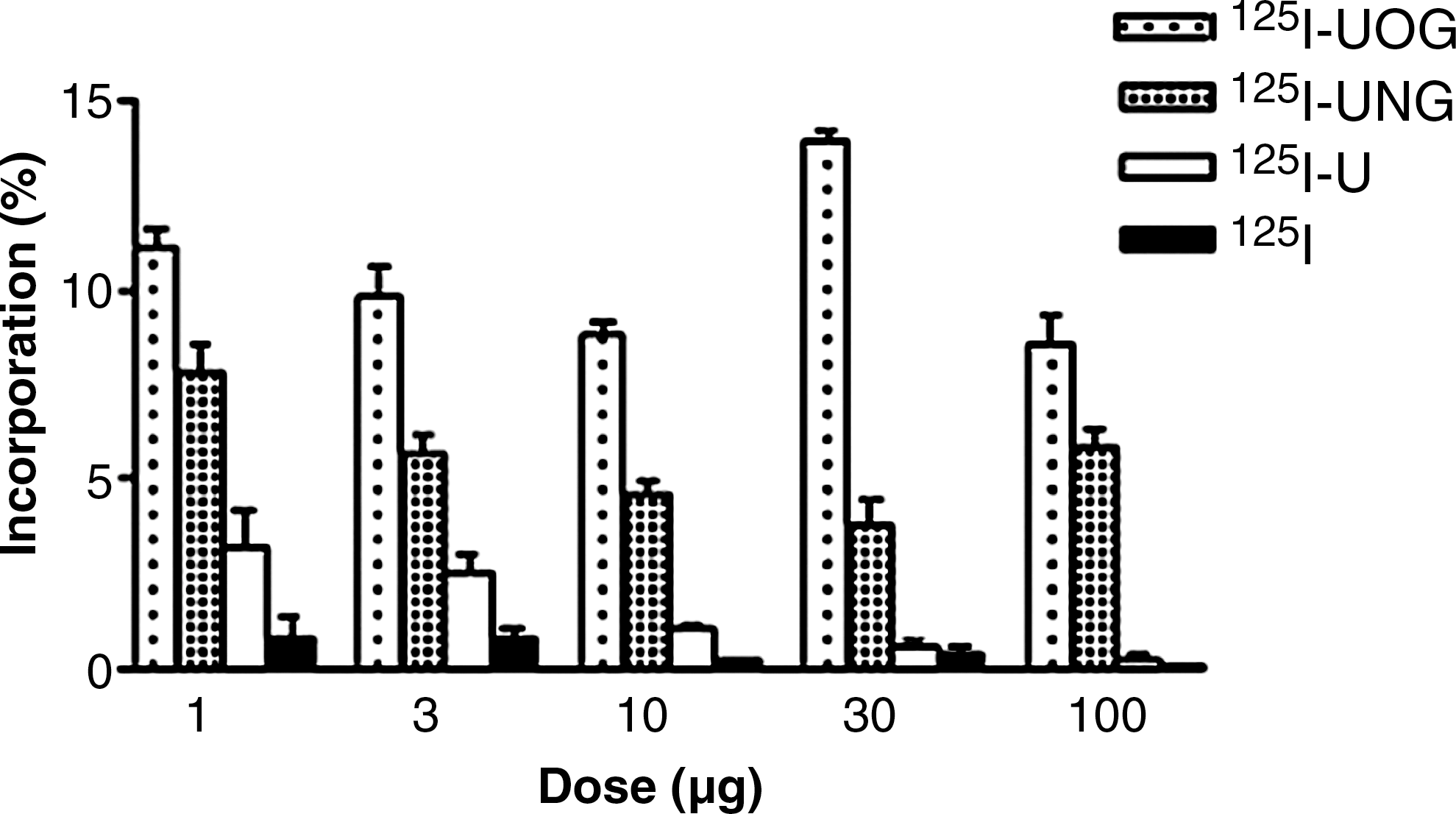
Dose-dependent incorporation values of I-125-labeled compounds. U, uracil; UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
The results show a significant difference between the incorporation of 125I-UOG, 125I-UNG, 125I-U, and 125I (Fig. 9). Incorporation values of the I-125-labeled compounds resembled each other on the Hutu-80, Caco-2, and Detroit 562 cells. Though both glucuronide derivatives had similar distributions on these cells, o-glucuronides had higher incorporation values. The incorporation of 125I-UOG was higher than that of other compounds on all cell lines (p < 0.05). The incorporation values of 125I-UNG on the three cell lines were three to four times smaller than those of 125I-UOG (p < 0.05). 125I-uracil and 125I had incorporation values below 1% on the three cell lines. The results show that the β-glucuronidase enzyme in determined cancer cells hydrolyzes o-glucuronides more than n-glucuronides.
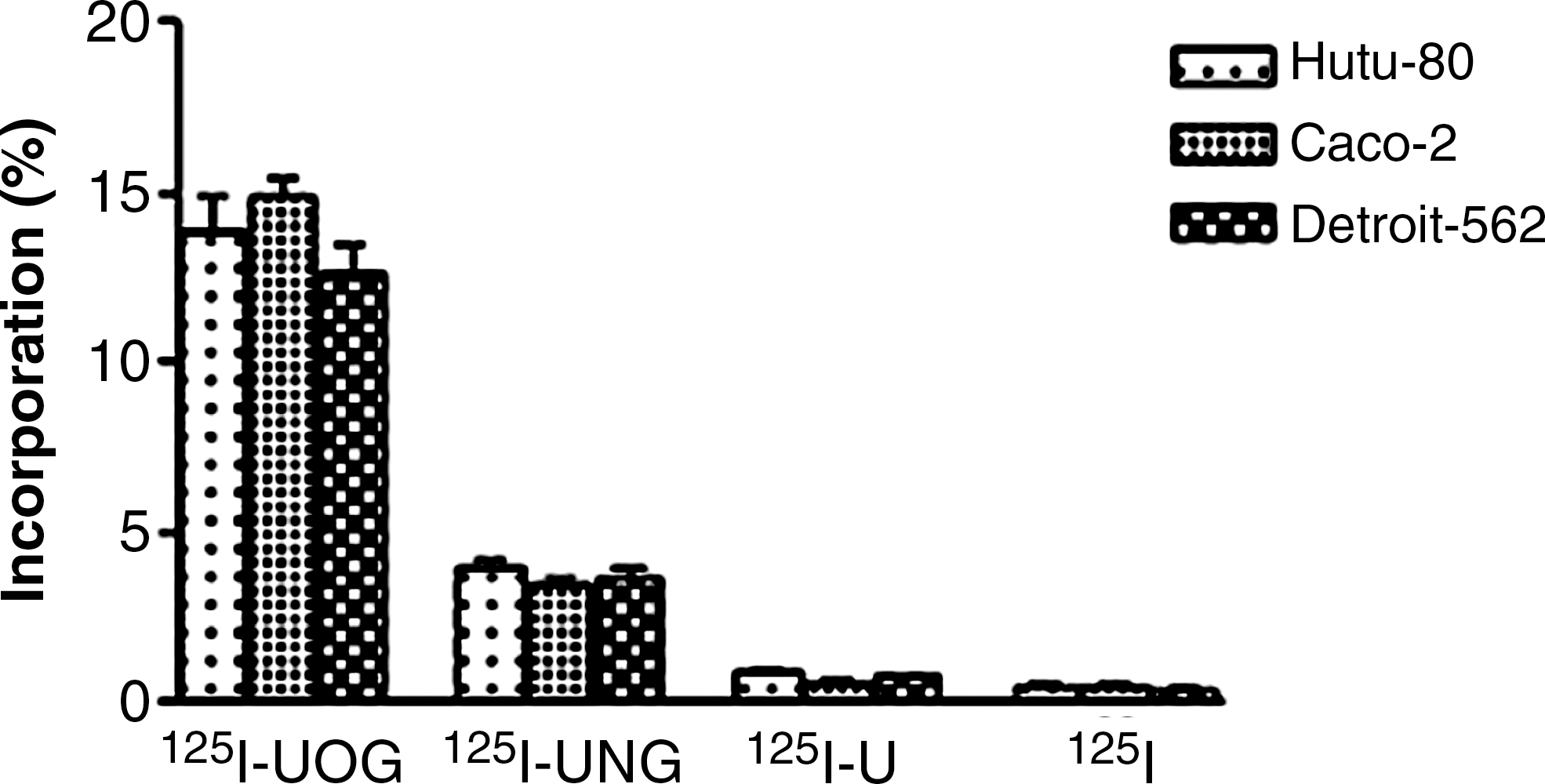
Incorporation of 125I-UOG, 125I-UNG, and 125I-uracil on adenocarcinoma cell lines. UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide; U, uracil.
The experiments were repeated with I-131 instead of I-125 to determine whether I-131 β-radiation caused damage to the cells. The incorporation of 131I-UOG was higher than that of other I-131-labeled compounds on all cell lines (p < 0.05), as seen in Figure 10. The incorporation values of I-125- and I-131-labeled compounds were compared and found to be similar for all cell lines. There was no statistically significant difference (p > 0.05) between 125I-UOG and 131I-UOG, 125I-UNG and 131I-UNG, 125I-uracil and 131I-uracil, and 125I and 131I.
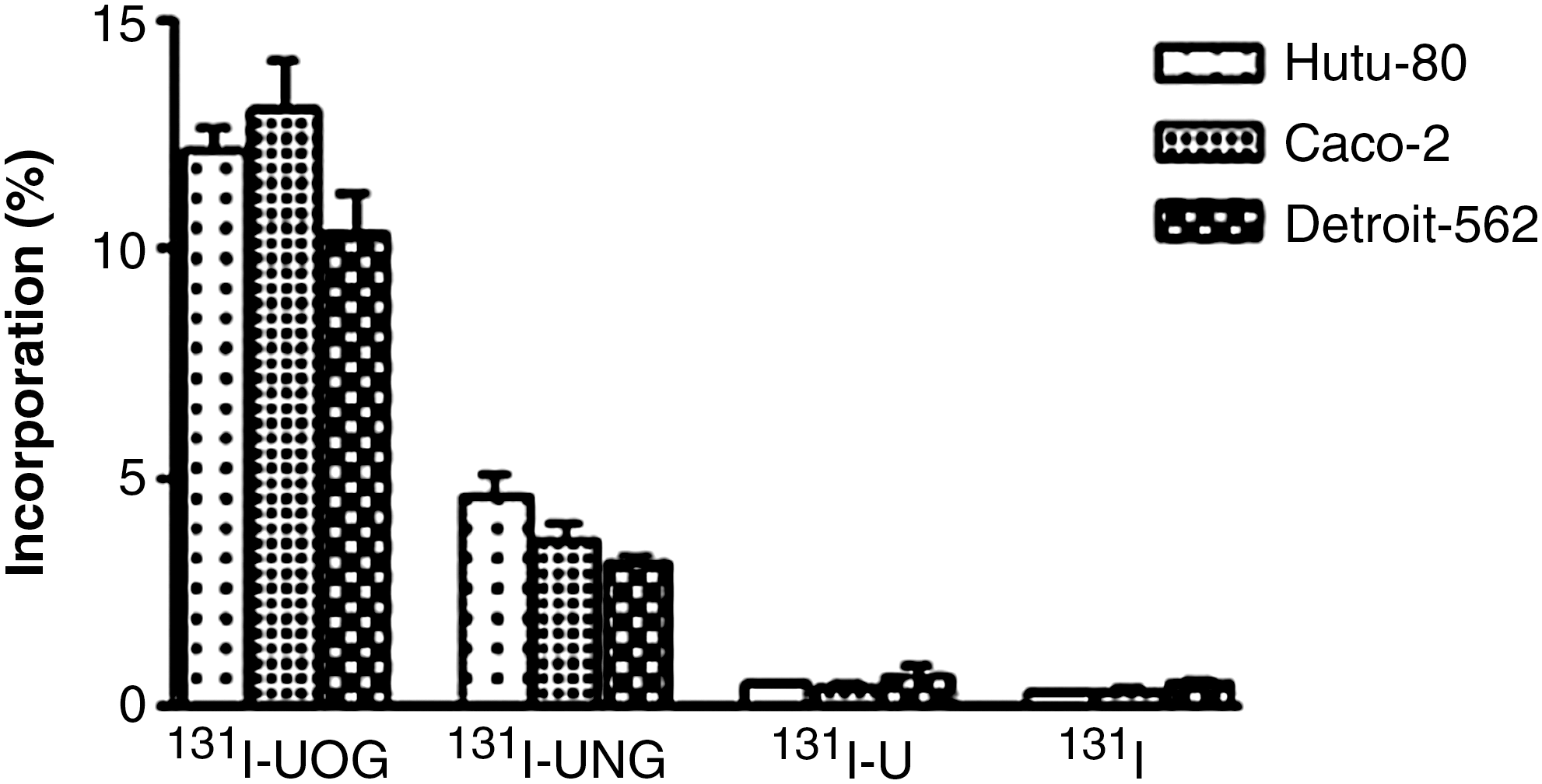
Incorporation of 131I-UOG, 131I-UNG, and 131I-uracil on adenocarcinoma cell lines. UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide; U, uracil.
Incorporations of I-125-labeled uracil glucuronides on normal cell lines (ACBRI 519) and adenocarcinoma cell lines (Hutu-80) were compared. 125I-UOG incorporated more than 125I-UNG, 125I-uracil, and 125I on both cell lines (Fig. 11). However, the incorporation of 125I-UOG on adenocarcinoma cell lines was higher than on normal cell lines, the difference being statistically significant (p < 0.05; Table 7). Incorporations of 125I-uracil and 125I on both cell lines were quite low. These results indicate that glucuronidation raises incorporation efficiency compared to normal cells. UOG was more selective toward adenocarcinoma cells.

Incorporation of I-125-labeled samples on normal cells. UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide; U, uracil.
UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide.
To study the radiation damage effect of increasing specific activity of 125/131I on the cells, UOG and UNG labeled with different specific activities of 125/131I were incorporated on Hutu-80 cells. The results are presented in Figure 12. Incorporation efficiencies of 125/131I-UOG and 125/131I-UNG were higher than those of 125/131I-uracil, I-125, and I-131. In all experiments, 125/131I-UOG showed higher incorporation than 125/131I-UNG. Although incorporation of I-125-labeled UOG and UNG did not change significantly by increasing the specific activity, incorporation of I-131-labeled UOG and UNG reduced with increasing specific activity because of radiation damage caused by I-131 decay.

Incorporation effect of increasing specific activity of 125/131I. UOG, uracil-o-glucuronide; UNG, uracil-n-glucuronide; U, uracil.
Discussion
The peaks identified with HPLC are due to tautomeric shifts of uracil between the amide and imidic acid forms at pH 7 (Figure 13). The lactam structure is the most common form of uracil. Therefore, the first and the second peaks were defined, respectively, as UOG and UNG, and theoretical lipophilicities confirmed that prediction. Ouhabi et al. 15 studied tautomeric forms of 2-amino-2-oxazoline heterocycle and reported that the amino form is more lipophilic than the imino form. The results of the present study agree well with this report. The n-glucuronide was more lipophilic and was therefore separated by reversed-phase HPLC. The n-glucuronide peak was behind the o-glucuronide peak in the reversed-phase HPLC.
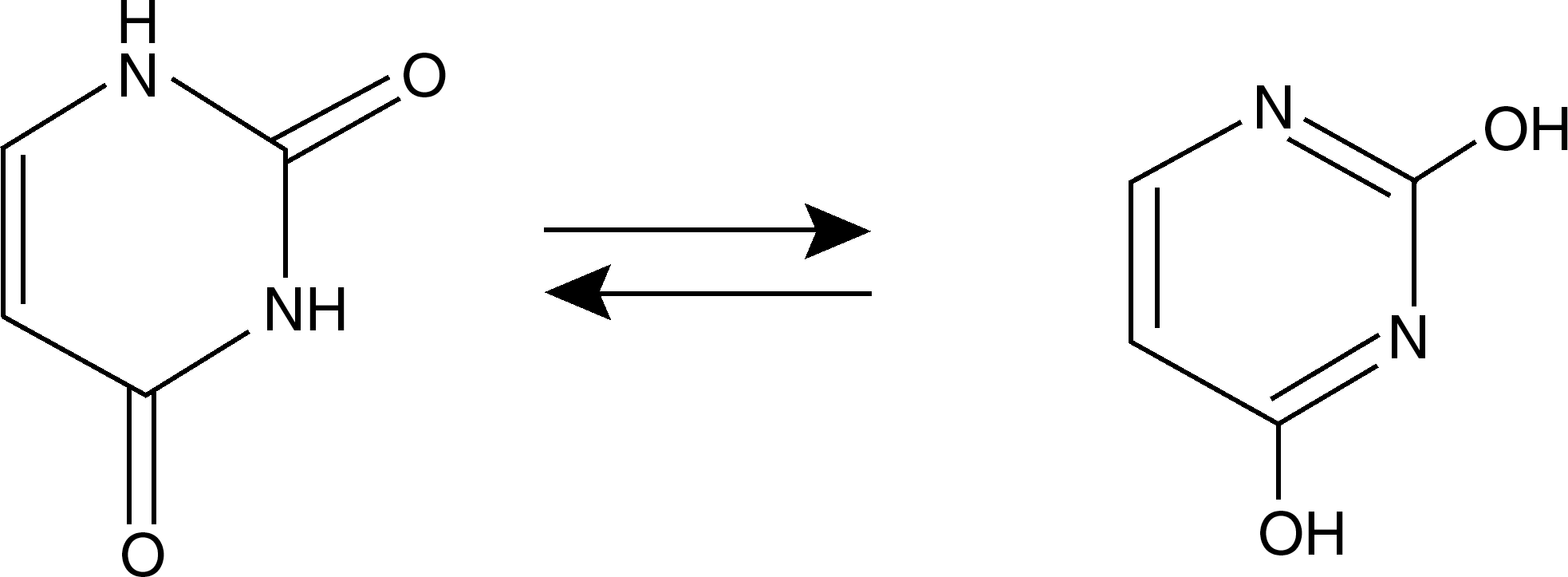
Tautomeric forms of uracil.
The lipophilicities for 131I-UOG, 131I-UNG, and 131I-uracil were − 5.05 ± 0.91, −2.13 ± 0.72, and +0.03 ± 0.02 (n = 5), respectively, which agreed well with theoretical values. Uracil was more lipophilic than its glucuronide derivatives because glucuronidation reduces lipophilicity.
From cell culture studies optimum incorporation time and dose were determined as 30 minutes and 30 μg/mL, respectively. Incorporation values of the I-125-labeled compounds on the Hutu-80, Caco-2, and Detroit 562 cells were similar. Both glucuronide derivatives had similar distributions on these cells, though o-glucuronides had higher incorporation values. It may be concluded that biological activities of the glucuronides may also differ. Giroud et al. 16 reported a number of studies on the in vivo effects of morphine 3-o-glucuronide and morphine 6-o-glucuronide, and concluded that M6G, with its remarkable analgesic activity, was more potent and longer acting than morphine when administered peripherally to animals and patients, while M3G seemed to be a weak antagonist of morphine.
Both 125I-UOG and 125I-UNG incorporated more with the three cell lines than did 125I-uracil and 125I. β-Glucuronidase localized in the ER may be responsible for the hydrolysis of drug glucuronides in cells. 17 The incorporation of o-glucuronide was about three times more than that of n-glucuronide, possibly because o-glucuronides hydrolyze more than n-glucuronides.
To exhibit the radiation damage effect of increasing the specific activity of 125/131I on the cells, incorporation of I-125- and I-131-labeled compounds on Hutu-80 cells were examined. Increasing I-131 activity caused more damage than increasing I-125 activity, owing to transportation of UOG and UNG to cells via ER. I-125 decays by electron capture, and is a very effective Auger electron emitter due to its specific decay characteristics. Two successive Auger cascades occur during each decay of I-125, consequently emitting on an average 21 Auger electrons in condensed matter per decay. These electrons have energies between 10 eV and 34 keV. 18 The range of Auger electrons emitted by an I-125 atom is about 40–45 nm, which is significantly short in condensed material. Thus, the local absorption of these electrons results in high-energy deposition in the decay vicinity of I-125. These electrons are very effective for cellular radiotoxicity if the 125I incorporates to DNA but not effective if it incorporates to the outer nucleus.
131I emits 182 keV per decay by several energy β-particles together with 364 keV energy gamma-ray photons. 19,20 Deposition sites in the cell are important in the selection of radionuclides. For example, if deposition occurs in the cell nucleus, an Auger electron emitter radionuclide may be better to kill the cell, while in the case of cell surface deposition a β-emitter may be preferable. The position effect may be vital in internal radiation damage. Hofer and Hughes studied mouse leukemia cells labeled with 125IUdR, 131IUdR, or 3HTdR for DNA targeting. They reported that the effect of 131I and 3H, both β-emitters, on cell killing was similar, while the decay of 125I was about 10 times more effective at the 37% survival level. In these experiments the dose–effect curves for the incorporated 131I or 3H had a shoulder in the low-dose region, which for 125I was linear, indicating a one-hit one-target interaction. 21 The results of the present study differ from that of Hofer and Hughes in that the cellular position of the glucuronides is not the cell nucleus but the ER. UDP-Glucuronosyltransferase (UGT) and β-glucuronidase enzymes exist in the ER and are responsible for the efficient inward transportation of glucuronides at the plasma membrane. 22 Therefore, longer-ranged 131I β-particles were more effective in damaging the cells than Auger electrons of 125I in this work.
Conclusions
Enzymatic glucuronidation using human UGT-rich microsome preparates extracted from Hutu-80 cells is an efficient method to prepare glucuronide derivatives. The products obtained can be used as prodrug molecules to carry more lipophylic and toxic drugs to β-glucuronide-rich targets. These glucuronide prodrugs may be deposited more in cancer cells than in healthy cells.
The β-glucuronidase enzyme hydrolyzes o-glucuronides more than n-glucuronides in cancer cells.
Radiolabeled glucuronide derivatives can be used in cancer therapy and tumor imaging depending on the properties of radioiodine for the β-glucuronidase-rich tissues, because glucuronidation leads to rapid and higher incorporation on adenocarcinoma cells.
Footnotes
Acknowledgments
This work was financially supported by the Scientific and Technological Research Council of Turkey (TUBITAK; contract no 105 S 486) and the Ege University Research Funds (contract nos. 06 NBE 03 and 07 ILAM 01). The authors thank Adnan Menderes University Science Technology Research and Application Center for helping with the cell culture experiments.
Disclosure Statement
No competing financial interests exist.
*
Zihnioglu F. Isolation, purification and immobilization of UDPglucuronyl transpherase. Ph.D. dissertation, Biochemistry Section, Chemistry Department, Graduate School of Natural and Applied Sciences, Ege University, Bornova, Izmir 1992;169.