
Abstract
Second mitochondria-derived activator of caspase (Smac) is a mitochondrial protein that promotes apoptosis in many kinds of cancers. Here, for the first time, the effects of Smac RNAi on growth and drug resistance to cisplatin [cis-diamminedichloroplatinum(II)] of lung cancer cells are investigated. Knockdown of Smac expression in A549 and 95D cells was mediated by transfection with pGC-FU vector containing siRNA sequences targeting human Smac with the lentivirus vector system. Smac was also overexpressed by transfection with pOE vector containing full-length coding region of Smac. Cell growth, cell cycle, and apoptosis were measured by methyl-thiazol tetrazolium assay, colony-formation assay, and flow cytometry. Drug resistance was performed by treatment with 10 μg/mL cisplatin. Downregulation of Smac enhanced cell growth and drug resistance to cisplatin of A549 and 95D cells, whereas overexpression of Smac did reversely. Smac helps inhibit cell growth and potentiate drug sensitivity to cisplatin of lung cancer cells.
Introduction
Lung cancer is the leading cause of cancer-related death, which accounts for over one million deaths worldwide per annual. 1 Approximately 80%–85% of all the lung cancers are non-small cell lung cancer, which include adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma. The disease is rarely curable, and the prognosis remains dismal at 15% of 5-year survival. 2 It is necessary to find new therapeutic molecular targets to ameliorate its outcome.
Second mitochondria-derived activator of caspase (Smac), also known as DIABLO (direct inhibitor of apoptosis-binding protein with low pI), is a proapoptogenic mitochondrial protein that stimulates apoptosis, possibly, by neutralizing inhibitors of apoptosis proteins (IAPs). 3 –5 Several studies have demonstrated that overexpression of Smac potentiates apoptotic death of many neoplastic cells, including Hodgkin lymphoma, 6 breast cancer, 7 glioblastoma, 8 and thyroid cancer. 9 Other studies have shown that the expression of Smac is reversely related to cancer progression in renal cell carcinoma, 10 testicular germ cell tumors, 11 colorectal cancer, 12 and lung cancer. 13 Although Sekimura et al. have shown that Smac mRNA transcripts decreased significantly during lung cancer progression, 13 there is no report about the effects of down- and up-regulation of Smac on growth and drug resistance of lung cancer cells.
Cisplatin or cis-diamminedichloroplatinum(II) (cDDP) is a platinum-based chemotherapy drug that is widely used for the treatment of a variety of tumors including small cell lung cancer. It acts in vivo, binding to and causing crosslinking of DNA, which ultimately triggers apoptosis. Cisplatin combination chemotherapy is the cornerstone of treatment of many cancers. However, unfortunately, the majority of patients with cancer who have received cisplatin will eventually relapse with cisplatin-resistant disease though the initial platinum responsiveness is usually high. Many mechanisms of cisplatin resistance have been proposed, including changes in cellular uptake and efflux of the drug, increased detoxification of the drug, increased DNA repair, and inhibition of apoptosis, 14 but much more remain unknown. It has been reported that downregulation of Smac contributes to chemotherapy resistance, whereas overexpression of Smac increases chemotherapy sensitivity including cisplatin sensitivity. 9,15 –17 However, there is no direct and specific evidence for the relationship between endogenous Smac expression and cisplatin resistance.
In this study, the effects of Smac down- and up-regulation on cell growth and drug resistance to cisplatin of two lung caner cell lines, A549 and 95D cells, have been investigated and this helps us gain more insight into the roles of Smac in lung cancer treatment and cisplatin sensitivity.
Materials and Methods
Cell culture
Human lung cell lines A549 (highly invasive human lung adenocarcinoma cell) and 95D (highly metastatic human lung carcinoma cell) were purchased from Cell Bank Type Culture Collection of Chinese Academy of Sciences (CBTCCCAS) and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (Gibco BRL) and penicillin (100 U/mL)/streptomycin (100 μg/mL) (Gibco BRL) at 37°C in humidified 5% CO2.
Smac RNAi and overexpression
A549 and 95D cells were transfected with lentivirus gene transfer vector containing siRNA sequences targeting Smac (pGC-FU vector for RNAi) or full-length coding region of human Smac (pOE vector for overexpression). pGC-FU vector containing siRNA with a green-fluorescent protein (GFP) sequence at the C-termination and the control vector were also transfected. The targeting sequence of the siRNA was 5′-GAGCTGAGATGACTTCAAA-3′. To verify the specificity of RNAi, a rescue experiment was performed. A GGGCAGAAATGACGTCTAA sequence, which was based on the synonymous codon replacement principle, was used to replace the interference sequence targeting SMAC in pOE vector, so that this rescue expression vector could make SMAC mRNA escape degradation. All the vectors were constructed by Genechem Co., Ltd (Shanghai, China) and confirmed by sequencing.
Cells were plated in 6-well plates (5 × 104 cells/well) until cell fusion reached 30%. Then, according to the MOI value (MOI = 20, number of lentiviruses: number of cells), appropriate amounts of lentiviruses were added to the cells. After 24 hours of transfection at 37°C, the medium was replaced by fresh DMEM medium and incubated for a further 48 hours. The efficiency of knockdown was tested by real-time RT-PCR and western blot analysis.
Real-time RT-PCR
After transfection for 5 days, total RNA was extracted from cells with Trizol (Life Technologies) and reverse-transcribed according to the manufacturer's instructions. About 5 ng of the cDNA was used for real-time PCR. The Smac gene was amplified with SYBR Master Mixture (Takara, Japan) on IQ5 (BioRad), comprised an initial denaturation at 95°C for 15 seconds, then 45 cycles at 95°C for 5 seconds, and 60°C for 30 seconds. The beta-actin gene was used as an internal control. PCR primer sequences for Smac gene were as follows: SMAC-F: TCAGAGATGGCAGCAGAAG; SMAC-R: CCCTCAATCCTCACGCAG.
Western blot
After transfection for 8 days, cells were lysed by precooled lysis buffer, and the protein content of the lysates was assessed. A 10% polyacrylamide gel was poured with a 5% stack. Then, 30 μg protein was loaded to each well, and gels were run at 30 mA for 2 hours. Transfer was semidry for 2 hours in standard transfer buffer. The resulting membrane was blocked in 5% nonfat dry milk blocking buffer and then probed with protein-specific antibodies overnight at 4°C. The membrane was then washed thrice with TBST, followed by incubation for 2 hours with appropriate secondary antibody at room temperature. After further washing, the membrane was developed using enhanced chemiluminescence (Amersham). Rabbit anti-Smac (Abcam; Cat log: ab13817) and mouse anti-GAPDH (Santa Cruz; Cat log: sc-32233) were used at a 1:200 and 1:6000 dilution, respectively. Secondary antibodies goat anti-rabbit IgG and goat anti-mouse IgG were used with the work concentration of 1:5000 and 1:6000, respectively (Santa Cruz; Cat log: sc-2005 and sc-2030).
Methyl-thiazol tetrazolium assay
Cell proliferation ability was assessed by methyl-thiazol tetrazolium (MTT) assay. Exponentially growing cells were incubated into 96-well plates with 2 × 103 cells per well, and after incubation for 24, 48, 72, and 96 hours, 10 μL of sterile MTT (5 mg/mL; Sigma-Aldrich Corp.) was added to each well. After a further incubation at 37°C for 4 hours, the reaction was stopped by adding 100 μL of dimethyl sulfoxide. After being thoroughly mixed for 10 minutes, the formazan production was determined by measurement of the spectrometric absorbance at 490 nm on an enzyme immunoassay analyzer (1420 multilabel counter).
5-Bromo-2′-deoxyuridine incorporation assay
The 5-bromo-2′-deoxyuridine Labeling and Detection Kit I (Roche) was used to measure cell proliferation. Control and Smac-overexpressing A549 cells were cultured in 96-well plates in 100 μL/well of RPMI-1640 medium until 70% confluence was reached. 5-Bromo-2′-deoxyuridine was then added to each well to a final concentration of 10 μM. After another incubation of 4 hours, cells were fixed, washed, and incubated with primary and secondary antibodies according to the manufacturer's instructions. The immune complexes were detected by the subsequent substrate reactions and quantitated by measuring the absorbance at 490 nm on an enzyme immunoassay analyzer (1420 multilabel counter).
Colony-formation assay
Exponentially growing cells of the parental, infected with Smac -RNAi-Lentivirus and negative control lentivirus were suspended in complete growth medium and seeded in 6-well plates with 200 cells per well. The plates were maintained at 37°C in a humidified incubator with 5% CO2 for 2 weeks. The visible colonies were subsequently recorded by a fluorescence microscope. After fixation in paraformaldehyde, the colonies were stained with giemsa for 10 min followed by taking pictures with a digital camera.
Cisplatin treatment
Two thousand tumor cells/well were cultured in 96-well microtiter plates in the presence/absence of 10 μg/mL cisplatin for 72 hours. Then, 10 μL of sterile MTT (5 mg/mL) was added to each well. After a further incubation at 37°C for 4 hours, the reaction was stopped by adding 100 μL of dimethyl sulfoxide; and the formazan production was determined by spectrometric absorbance at 490 nm on an enzyme immunoassay analyzer (1420 multilabel counter).
Flow cytometry assay
After treatment with 10 μg/mL cDDP for 72 hours as mentioned above, cells in each well were harvested and cell apoptosis was determined using an Annexin V FITC Apoptosis Detection Kit (Calbiochem). Tests were performed in triplicate for each sample, and analyses were performed by FAC-Scan Flow Cytometer (Becton Dickinson, USA) in accordance with the manufacturer's guidelines.
Statistical analysis
All values in the text and figures are expressed as the mean ± SEM. Statistical significance was determined by a Student's t-test using the software package SPSS (
Results
Knockdown of Smac expression by RNA interference
A lentiviral vector system was used to express siRNA against Smac with GFP as a reporter. To determine whether the recombinant vectors were transfected to human lung cancer cells, A549 cells infected with Smac-RNAi-Lentivirus were assessed under a fluorescence microscope. After transfection with the encoding lentivirus for 72 hours, more than 80% of the A549 cells expressed GFP (Fig. 1A), indicating a high and stable transfection system. Real-time PCR and western blot results confirmed that transfection with Smac-RNAi-Lentivirus significantly decreased the expression of Smac at mRNA and protein levels (Fig. 1B, C).

Knockdown of second mitochondria-derived activator of caspase (Smac) expression by RNAi.
Smac RNAi promotes proliferation and growth of lung cancer cells
To assess the effect of Smac knockdown on proliferation and growth of human lung cancer cell lines A549 and 95D, MTT assay and colony-forming assay were performed. The growth curves presented in Figure 2A and B revealed that pKD-transfected cells had a significant increase in proliferation as compared with that of pNC-transfected cells in both cell lines, which indicates that knockdown of Smac promotes the proliferation ability of human lung cancer cells. In the rescue experiment, the phenotype of cell growth promotion by RNAi could be rescued by reoverexpression of SMAC protein, suggesting that the SMAC RNAi was specific. In addition, the results from colony-forming assay showed that the colony amounts and size of pKD-transfected cells had an obvious increase on comparison with pNC-transfected cells (Fig. 2C, D), thus providing evidence that Smac knockdown significantly promoted colony formation of lung cancer cell A549.
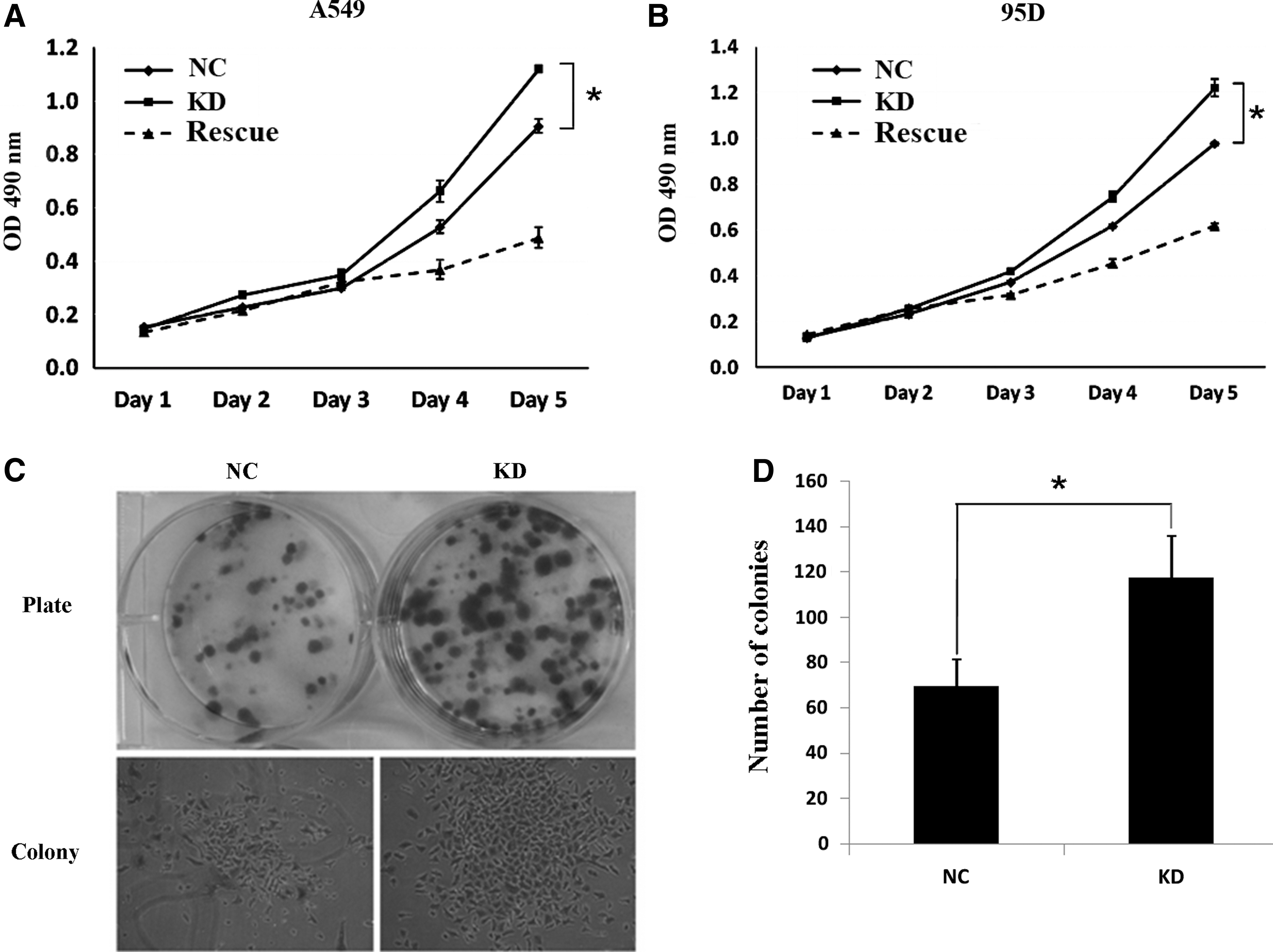
Kncokdown of Smac promotes lung cancer cell growth.
Smac RNAi enhances the drug resistance of lung cancer cells to cisplatin
Cisplatin is one of the most widely used anticancer drugs with genotoxicity. 17 It can also induce apoptotic cell death. 18,19 When grown in the absence of cisplatin (cDDP−), the growth of pKD-transfected A549 and 95D cells was quite similar to that of pNC-transfected cells. However, when treated with 10 μg/mL cisplatin (cDDP+), pKD-transfected cells exhibited significantly enhanced resistance to cisplatin as compared with pNC-transfected cells (Fig. 3A, B), which revealed the induction of drug resistance to cisplatin by Smac downregulation. Further, flow cytometry showed that the apoptosis induced by cisplatin was also inhibited by Smac knockdown. In both lung cancer cells lines, the proportion of cells at early and late stages of apoptosis (LR and UR) was largely reduced when Smac was downregulated, whereas, at the same time, the proportion of living cells was dramatically increased (Fig. 3C, D).

Knockdown of Smac enhances cisplatin resistance of lung cancer cells.
Smac overexpression inhibits cell growth and increases cisplatin sensitivity of A549 cells
To reconfirm the stimulating effects of Smac downregulation on lung cancer cell growth, A549 cells overexpressing Smac were established. As shown in Figure 4A, protein content of Smac was greatly increased in pOE-transfected A549 cells, which validated the effectiveness of lentivirus-mediated Smac overexpression. In contrast to Smac knockdown, overexpression of Smac significantly inhibited cell growth (Fig. 4B, C) and increased cisplatin sensitivity of A549 cells (Fig. 4D). These data demonstrated the important roles of Smac in cell growth and drug sensitivity of lung cancer cells from another aspect. See appendix for Supplementary figures showing more information on these results.

Overexpression of Smac inhibits growth and cisplatin resistance of A549 cells. Overexpresion of Smac (OE) was confirmed by western blot
Discussion
Every year, lung cancer causes more than a million deaths all over the world. Traditionally, lung cancers are commonly treated by surgery, chemotherapy, and radiation therapy. Although great improvements have been achieved in the past decades, the outcomes and 5-year survival remain dismal. 2 New therapies with high specificity and efficiency are eagerly needed.
The Smac is a recently identified proapoptotic protein that inhibits IAPs, leading to TNF-receptor mediated apoptosis. 20,21 Overexpression or mimics application of Smac confers hypersensitivity to apoptotic death of many cancer cells. 7,22 –26 Many studies have shown that there are clear inverse correlations between Smac expression and progression in many kinds of cancers. 9,10,12,27,28 Recently, Sekimura et al. reported that Smac mRNA transcripts significantly decreased during lung cancer progression, suggesting that Smac is a novel prognostic marker in lung cancer and may play a role in the carcinogeesis, progression, and prognosis of primary lung cancer. 13 In addition, it has been reported that Smac release was required for cisplatin-induced apoptosis and the expression level of Smac was positive related to chemotherapy sensitivity. 29 Downregulation of Smac contributes to chemotherapy resistance, whereas elevated Smac expression by transfection rendered resistant cancer cells sensitive to cisplatin-mediated cytotoxicity. 10,16 However, there is scarce direct evidence illustrating the effects of altered Smac expression on lung cancer growth and on cisplatin resistance up to now.
In our study, the effects of Smac down- and up-regulation on lung cell growth and drug resistance to cisplatin were tested. In two lung cancer cell lines, A549 and 95D, knockdown of Smac by siRNA significantly stimulated cell proliferation and colony formation (Fig. 2). Lung cancer cells showed enhanced resistance to cisplatin-induced cell death when endogenous Smac expression was downregulated (Fig. 3A, B). Cisplatin-induced apoptosis was also greatly suppressed when Smac expression was inhibited (Fig. 3C, D). These results were in high accordance with previous reports about the proapoptotic effects of Smac on cancer cells. To ensure this conclusion, the results were reevaluated by overexpressing Smac in A549 cells. In contrast with Smac knockdown, Smac overexpression remarkably inhibited cell proliferation (Fig. 4B) and colony formation (Fig. 4C) and sensitized A549 cells to cisplatin-induced cell death (Fig. 4D). These data reconfirmed the inhibitive effects of Smac on lung cancer cells.
It has been well documented that the N-terminal of SMAC binds to IAPs and neutralizes them, thus stimulating apoptosis. 3 –5 Some apoptosis inhibitors such as fibroblast growth factor 2 can suppress apoptosis by blocking the release of SMAC from mitochondria to the cytoplasm. 30 Therefore, the expression level of SMAC is critical for apoptosis induction. Knockdown of SMAC by siRNA in lung cancer cells might decrease the neutralization of IAPs, leading to a higher amount of functional IAPs and inhibiting the apoptosis. The decrease of cellular SMAC may also enhance the function of fibroblast growth factor 2 (a kind of apoptosis inhibitor), thus blocking apoptosis of lung cancer cells. On the other hand, overexpression of SMAC would neutralize more IAPs and inhibit the function of some apoptosis inhibitor, thus significantly enhancing apoptosis of lung cancer cells.
Due to the importance of Smac in determining the sensitivity of cancer cells to apoptotic death, it is widely accepted that Smac is a useful target for the development of more effective chemopreventive strategies and agents. 23,31 A lot of small molecule Smac mimetics are being developed as a novel class of anticancer drugs. 31,32 Our results provided robust evidence for the important roles of Smac in suppressing lung cancer cells growth and sensitizing them to chemotherapeutic drugs. Therefore, Smac is also a promising target for the development of effective drugs for lung cancer treatment.
Footnotes
Acknowledgments
The authors are thankful for the financial support from National Natural Science Foundation of China (No. 30973473).
Disclosure Statement
No competing financial interests exist.