
Abstract
Computational chemistry is playing an increasingly important role in drug design and discovery, structural biology, and quantitative structure–activity relationship studies. A series of 4(3H)-quinozolone derivatives were screened for two-dimensional quantitative structure–activity relationship studies and subsequently their absorption, distribution, metabolism, and excretion (ADME) properties with the use of soft modeling techniques after selecting suitable descriptors for molecular structure. Multiple linear regression analysis was performed for this study. The final quantitative structure–property relationship mathematical models were found as follows:
Equation
Introduction
Protein tyrosine kinases are enzymes involved in many cellular processes such as cell proliferation, metabolism, survival, and apoptosis. Several protein tyrosine kinases are known to be activated in cancer cells and to drive tumor growth and progression. Unregulated activation of these enzymes, through mechanisms such as point mutations or overexpression, can lead to a large percentage of clinical cancers. 1,2
Inhibitors of tyrosine kinase, which work as a new kind of effective anticancer drug, are important mediators of cellular signal transduction that effect growth factors and oncogenes on cell proliferation. 3,4 Blocking tyrosine kinase activity therefore represents a rational approach to cancer therapy. Epidermal growth factor receptor (EGFR), which plays a vital role as a regulator of cell growth, is one of the intensively studied tyrosine kinase targets of inhibitors. EGFR is overexpressed in numerous tumors, including those derived from brain, lung, bladder, colon, breast, head, and neck. As the hyperactivation of EGFR has been associated with these diseases, inhibitor of EGFR has potential therapeutic value and it has been extensively studied in the pharmaceutical industry.
One could not, however, confirm that the compounds designed would always possess good inhibitory activity to EGFR, and also the experimental assessments of inhibitory activity of these compounds are time-consuming and expensive. Consequently, it is of interest to develop a prediction method for biological activities before the synthesis. Quantitative structure–activity relationship (QSAR) information relating chemical structure to biological and other activities was searched by developing a QSAR model. Using such an approach one could predict the activities of newly designed compounds before a decision is being made whether these compounds should be really synthesized and tested.
QSAR analysis is the analysis of the quantitative relationship between the experimental activity of a set of compounds and their structural properties using statistical methods. 5 –9 The experimental information may associate with biological properties such as activity, toxicity, or bioavailability, which are taken as dependent variables in building a model. The parameters to be calculated are numerous descriptors that are indicative of molecular structures. In QSAR, the number of compounds with the biological activity values available is usually small compared with the number of structural descriptors.
In the same direction, and in continuing effort to find more potent selective lead compounds, the present study describes the design of a series of 4(3H)-quinozolone derivatives as possible antitumor agents that may act through EGFR inhibition as per a previous work. 10 –13
For the ease of multiple linear regression (MLR) implementation and the interpretability of the resulting equations, MLR techniques were used for building QSAR models. In the present work, modified ACO algorithm was employed for variable selection in MLR analysis of EGFR inhibitory activity and compared with electrical network analysis (ENA).
Materials and Methods
Computational details (QSAR study)
QSAR studies were performed on a series of quinazolone derivatives. For this purpose, various molecular properties were calculated and these were correlated with the biological activity to obtain various QSAR models.
A model may have one or more outputs and these may be descriptors, physical properties, or dependent variables. The descriptors mentioned fall into two categories: quantum mechanical and fast descriptors. The quantum mechanical descriptors were calculated using the PM3 semiempirical method. Fast descriptors are a set of efficient algorithms that can be used to calculate a variety of two-dimensional molecular properties. These are further classified into topological descriptors, structural descriptors, thermodynamic descriptors, information-content descriptors, and electro topological descriptors. κ(1), Wiener (ω), and χ(3) descriptors fall into the topological class of fast descriptors, which are two-dimensional descriptors based on graph theory concepts. These indices have been widely used in quantitative structure–property relationship and QSAR studies. They help to differentiate molecules according to their size, degree of branching, flexibility, and overall shape.
The Wiener index is the sum of the chemical bonds existing between all pairs of heavy atoms in the molecule. The κ(1) and χ(3) indices fall into Kier and Hall molecular connectivity indices. χ(3) emphasizes the different aspects of atom connectivity within a molecule. Basically, it explains the influence of clustering in the compound on the activity. κ(1) is a Kier shape index that intends to understand the different aspects of molecular shape.
Results
The chemical structures and the minimum inhibitory concentrations (MIC, in μg/mL) of 19 quinazolone derivatives are given in Table 1, which was calculated as per a previous work. 9 –12
MIC, minimum inhibitory concentration.
The data in Table 1 reveal a large variation in the activities. Four of the 15 compounds showed potent activity (MIC: 1–5 μg/mL), 6 were less potent (MIC: 15 μg/mL), and the rest were practically inactive. As the object of designing new drugs is achieving low MIC values, so that the drug may be administered in low dosage and the problems of drug resistance are minimized, this study looked at the various structure-related properties of the active compounds. Table 1 shows that short aliphatic chains lead to higher activity. Branching of the alkyl chain also does not favor activity, as revealed by the data.
To introduce a quantitative aspect to these conclusions, various structure-related parameters for each of the molecules were calculated and a quantitative structure–activity analysis of the data was performed. Based on this, we have calculated ADME and biological activity (Fig. 1). The number of compounds was limited to only 15. Therefore, a genetic function approximation (GFA) was applied to reveal the important parameters affecting the activity. Moreover, the data on MIC do not follow a normal distribution. Accordingly, the data were transformed to their logarithms.
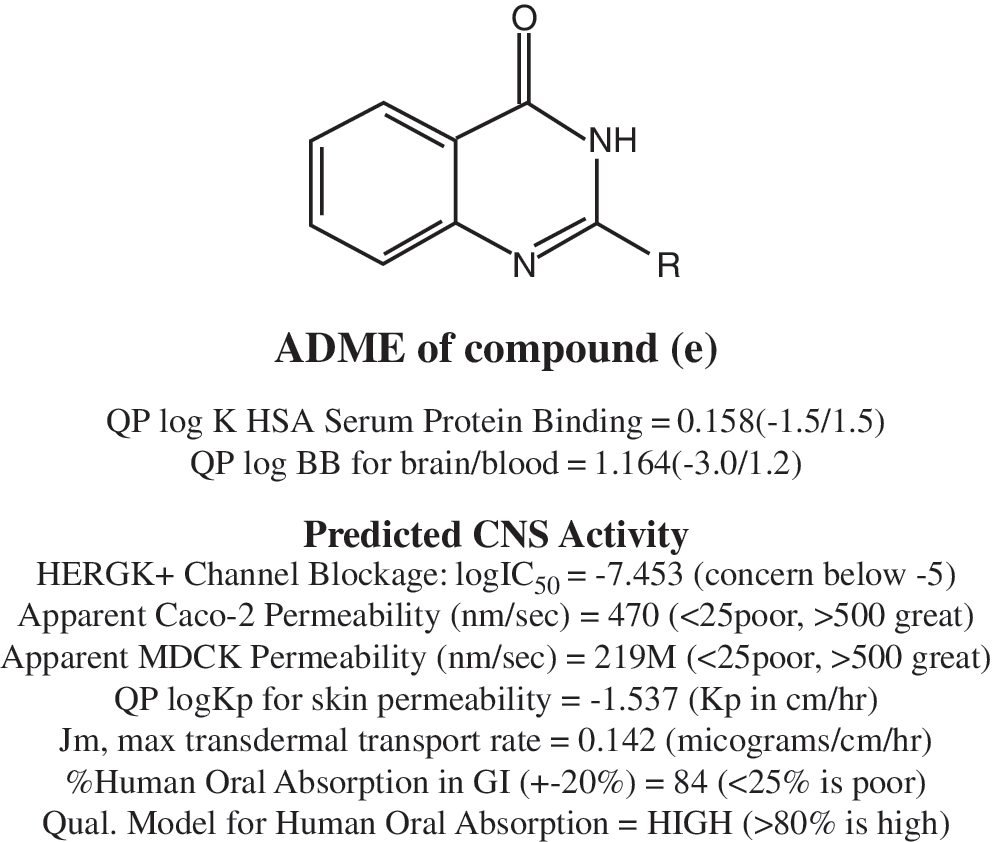
Substituted 4(3H)-quinozolone and predicted ADME. CNS, central nervous system.
The GFA method, implemented in regression analysis, was employed for selecting the “best” regression models, and these are given in Table 2. The “goodness of fit” of the models was tested on the basis of the squared correlation coefficient (R 2). For testing the predictive performance of the models, R 2 CV Leave One Out, the squared cross-validated coefficient method, was used. The Leave One Out approach consists in developing a number of models with one sample omitted at a time. After developing each model, the omitted data were predicted and the differences between the experimental and predicted activity values were calculated and plotted (Fig. 2). The five best models that were produced are shown in Table 2.

Predicted and experimental inhibitory activity. MIC, minimum inhibitory concentration.
Discussion
Among the models shown in Table 2, the best is Model 1 with R 2 = 0.862. This is a penta-parametric regression equation. Interestingly, Model 1 also shows the best predictive power with R 2 CV = 0.693. Model 2, with a slightly smaller correlation coefficient, is essentially the same as Model 1. According to Model 1, log(1/MIC) increases directly with κ(1), but it is inversely related to χ(3), total dipole moment and its x component, and the Coulson charge on N3. In other words, the MIC increases with increase in χ(3), μ T, μx , and q N, but decreases with increase in κ(1). Similarly, the other four model equations also contain five descriptors, for example, Model 2, which behaves almost similar to Model 1, is inferior to it in statistical characteristics, both in terms of R 2 and R 2 CV (R 2 = 0.835, R 2 CV = 0.672). Model 3 uses the Wiener index (ω) instead of κ(1), but the other descriptors remain the same as in the above models. However, the Wiener index does not bring much difference in terms of statistical characteristics, with R 2 = 0.828, and R 2 CV = 0.646. Model 4 replaces two descriptors, namely the κ(1) and total dipole moment, of Model 1 with new descriptors, that is, the total energy and electrostatic charge on the carbons carrying the substituent R. In comparison to Model 1, Model 4 displays both poor “goodness of fit” as well as predictive power, as suggested by the values, R 2 = 0.815 and R 2 CV = 0.628. Model 5 makes replacement of total dipole moment and its x component with the octapole moments in xxx and xyy dimensions.
Using Model 1, the predicted inhibition constants of the compounds are presented in Figure 1 and Table 1. Of all the molecular (topological, thermodynamical, and structural) descriptors derived from the calculations based on GFA model of regression analysis, the important influential molecular descriptors for the 15 quinazolone derivatives and their experimental and predicted MIC values are presented in Table 1. These were descriptors used to select the dominant parameters affecting the inhibitory activity of the compounds.
In the QSAR model, the following properties appear in the topmost equations: χ(3) cluster, κ(1), Wiener index, Coulson charge on N3, electrostatic charge on C2, dipole moment (x), total dipole, octapole moment, and total energy. This list indicates that structural (topological) as well as electronic factors contribute to the activity or inactivity of a given compound. However, a deeper introspection of the actual quantitative effect of these parameters on the activity value is required. Deciphering the information available from a QSAR model needs the study of coefficients of these properties as they appear in the top equations.
χ(3) cluster contributes negatively toward activity, which leads to less clustering in the side chains. κ(1) has a positive coefficient, though of a comparatively lower value. It signifies that contacts of first degree between atoms are beneficial in improving the activity or that branching is not a favorable trait. Clustering could result in bad grades. Very long chains are also not recommended as elongation of the side chain has no major effect on the electronic contribution toward activity. These points motivated to choose simple 2–3 carbon atom chains to be introduced near the quinazolone moiety.
Conclusions
The results given above indicate that QSAR of MICs of quinazolone derivatives to EGFR can be modeled with a few molecular descriptors. The best model is a penta-parametric regression equation with good statistical fit and good predictive values. An analysis of the descriptors that are involved in the models indicates that the MIC is influenced largely by the Coulson charge on N3 and the electrostatic charge on C2. Another descriptor that influences significantly is χ(3), which is an indicator of clustering in the compound. Other less-significant descriptors are κ(1), total dipole moment, and its x component. Thus, the analysis indicates good correlation between structure and activity. In short, the MIC of derivatives can be improved (i.e., decreased) by including structural features that will decrease χ(3), Coulson charge of N3, electrostatic units (ESU) charge of C2, dipole moment, and its x component, while increase the value of the κ(1) descriptor.
Footnotes
Disclosure Statement
No financial conflicts exist.