
Abstract
The need to monitor cancer therapy-induced cellular and tissue changes using noninvasive imaging techniques continues to stimulate both basic and clinical research. Monitoring changes in cellular proliferative capacity that occur after treatment with radiation and/or chemotherapy has the potential to provide longitudinal information on the cellular dynamics of tumors before, during, and after therapeutic intervention. Cells can lose their reproductive potential through one of several mechanisms, including apoptosis and autophagy (which are forms of programmed cell death), premature senescence, or necrosis. When a tumor responds to therapy, current imaging methods do not provide information about the exact mechanism of cell death executed. We are now beginning to develop the molecular imaging tools that will enable us to noninvasively image cell death mechanisms both in experimental models and in the clinical cancer environment. Studies with these imaging tools will contribute to a better understanding of therapeutic responses and assist in the design and evaluation of more effective treatments. This review examines the state-of-the-art in the use of (radio)tracers for the purpose of imaging mechanisms of tumor cell inactivation (cell death) in animal models and in clinical trials.
Introduction
Effective radiation therapy (XRT) or chemotherapy will inactivate cancer cells either through programmed (apoptosis and autophagy) or uncontrolled (necrosis) cell death or through the induction of cellular premature senescence. Apoptosis is a well-studied tumor response to therapy; however, other mechanisms of tumor cell death are also induced after treatment. For instance, it is known that necrosis occurs in some tumor types and, whereas necrotic cell death is usually characterized as uncontrolled, there is now accumulating evidence that, in some cases, cellular events and cascades in necrosis represent a more programmed course of events. 1 Research has also highlighted the importance of cell senescence and autophagy after treatment with cytotoxic agents. Cells are able to use these various mechanisms to respond to cell stress. The interplay between modes of cell death and how they are connected is complex. 2 –4 The type of cell death executed in a particular system depends on factors such as the cell type, genotype, microenvironment, whether cells are adherent, and the dose and nature of the cytotoxic agent. A detailed account of the biology of cell death mechanisms is beyond the scope of this review, but the general characteristics of each mechanism are outlined in Table 1 and a review on cell death mechanisms in tumors has been presented by Okada and Mak. 5 Our understanding of these processes is predominantly derived from studies on cells in culture, and, at present, there are very limited options to probe these processes in solid tumors before and after therapy.
Modified from Okada and Mak. 5
TUNEL, terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling; SA-ß-gal, senescence-associated ß-galactosidase; LC3-II, microtubule-associated protein light chain 3-II.
Radiopharmaceutical-based nuclear medicine techniques have the potential to image cell death mechanisms at the cellular level due to their very high sensitivity. The science of radiopharmaceutical design is such that it is often possible to rationally design an appropriate ligand once a suitable target has been identified. Imaging of the various modes of cell death relies on (1) the identification of cellular targets that are unique to that process and (2) the development of radiolabeled probes that will bind with high selectivity and specificity to that target. Unfortunately, no single generic radiotracer is capable of imaging the abovementioned four very different and complex mechanisms that result in tumor cell death. This is because there is no single cellular target that is common to all cell death mechanisms and yet sufficiently unique to identify cell death from other biological processes. The induction of cell death by the mechanism of apoptosis is a desired effect of chemotherapy and radiotherapy. If the treatment does not induce apoptosis, treatment may be inducing necrosis, which could lead to undesirable inflammatory effects, or treatment may invoke a senescence response. At a preclinical level, probing the individual mechanisms of cell death will provide details of cellular response to novel therapies, whereas, at the clinical level, the physician will be better positioned to use this information to make informed decisions about patient care, such as changing a course of therapy that is not inducing a desired cell-death response. Multiple probes will be required to effectively determine the mechanisms of cell death, and this realization has resulted in both preclinical and clinical investigations with imaging radiotracers.
In a clinical context, imaging cell death in vivo has great significance in evaluating therapeutic response and has the potential to assist in the design of appropriate individualized and optimized therapy. Conventional imaging modalities (e.g., computed tomography [CT] and magnetic resonance imaging [MRI]) typically provide anatomical information that may not indicate tumor response to therapy until 6–12 weeks after treatment initiation. With conventional imaging modalities, ineffective therapies may be used for long periods of time before these treatments' ineffectiveness becomes evident. Evaluation of tumor response to chemotherapy and external-beam radiotherapy (XRT) after the earliest cycles of therapy would allow optimization of dosing and therapies, thereby improving patient outcomes and quality of life. Advantages of imaging with radiotracers include (1) noninvasiveness; (2) the ability to detect lesions earlier than X-ray–based or MRI imaging modalities; and (3) the fact that various parameters can be imaged with different probes. There are a number of tracers at various stages of development as possible probes for imaging cell death, which are discussed in this article (Table 2).
Discussed in this review.
PS, phosphatidylserine; ODN, oligonucleotide; 18FDG, fluorodeoxyglucose.
Imaging Tumors and Tumor Response to Therapy with Conventional Radiotracers
We are able to measure metabolic activity of tumors using the positron emission tomography (PET) radiopharmaceutical 18F-fluorodeoxyglucose (18F-FDG), in which the uptake of this agent is tied to glucose transport and utilization. 18F-FDG is an excellent probe for many tumors, with its primary applications being in the detection of metastatic disease, the staging of primary tumors, and in the follow-up to assess residual or recurrent disease. Some cancers, such as prostate, thyroid, testicular, renal, and bladder cancers, have variable or low uptake of 18F-FDG that limits the use of 18F-FDG PET in staging of these diseases and makes 18F-FDG a poor candidate for assessing early tumor response. 6 Furthermore, given that inflammatory cells, which are often present after therapy, have a high uptake of 18F-FDG, this tracer is generally not used for the early assessment of tumor viability. The ability of 18F-FDG to predict treatment outcome may be dependent on tumor type or on the measured parameter (i.e., standardized uptake value or metabolic tumor volume). 7,8 18F-FDG PET does not provide direct information on cell death and only provides an indirect measurement of tumor response. Given that biochemical responses of cells to chemotherapy and XRT occur within the first few days, there has been a vigorous research effort to develop molecular imaging tracers that will be able to assess specific features of cell death within 1–3 days.
18F-FDG PET is approved for monitoring response to treatment in breast cancer. 6 After one cycle of chemotherapy, responding tumors indicated a decline in standardized uptake value of 18F-FDG. 6 Tumor cells are also capable of changing their energy source to survive. 9 For example, some breast tumors can use fructose as an energy source, which decreases the usefulness of 18F-FDG PET as an imaging tool and minimizing its value for assessing tumor response to therapy. 10
Apoptosis is an energy-dependent process, and it may logically be considered that successful treatment will induce apoptosis and a subsequent increase in glucose (or 18F-FDG) uptake. This is known as a flare effect of tumor uptake. 11 The flare effect is not well- studied and may not be a universal effect in all tumors. However, it is advised that patients with breast cancer wait after chemotherapy for a minimum of 10 days before an 18F-FDG PET scan 11 to avoid this flaring effect and to avoid increased uptake due to inflammatory response. The wait time indicated for other tumor types is even longer. Guidelines for lymphoma recommend 3 weeks between the last chemotherapy treatment and 18F-FDG PET, and after XRT, it is recommended to wait 8–12 weeks before 18F-FDG PET. 11 It is clear that 18F-FDG has serious limitations in timely and effective imaging of cell death and tumor response to therapy.
Apoptosis Molecular Imaging
Apoptosis is a genetically controlled process (i.e., a form of programmed cell death) that is characterized by distinct biochemical and morphological changes including nuclear condensation, phosphatidylserine (PS) externalization, cytoplasm shrinkage, nonrandom DNA degradation, plasma membrane blebbing, and fragmentation of the cell into small apoptotic bodies. 12 Apoptosis is mediated by both intrinsic 13 and extrinsic 14 pathways that result in activation of a biochemical cascade in which cysteine-aspartic proteases (caspases) function as either initiators (caspases 2, 8, 9, and 10) or executioners (caspases 3, 6, and 7). The deregulation and suppression of apoptosis in tumors results in increased proliferative capacity and resistance to therapy. Tumor response to XRT is poorly understood and the role of apoptosis as a cell death mechanism after XRT is not well defined. 5,15 Therefore, the in vivo imaging of apoptosis with a radiopharmaceutical would be of great value in understanding the role and prevalence of apoptosis after XRT and chemotherapy. The changes in cell structure and biochemical processes during apoptosis provide attractive targets for molecular imaging. PS externalization and caspase activation have been the main cellular processes investigated as targets for molecular imaging. PS, which is ordinarily restricted to the inner layer of the phospholipid membrane, is externalized early in the apoptotic process. 16,17 This has led to the development of imaging probes for apoptosis that bind to externalized PS, as discussed below, and probes targeting activated caspases.
Caspase Targeted Probes
A target for imaging apoptosis is the presence of activated executioner caspases 3, 6, and 7. 18 Most caspase-directed probes bind directly to activated caspases and act as inhibitors. Current challenges of caspase-directed probes include efficient cell entry and cellular accumulation. The caspase inhibitor WC-II-98 has been radiolabeled with 18F and 11C and used with PET to image caspase-3 activation in tissues undergoing apoptosis. 18,19 WC-II-98 is a nonpeptide-based isatin sulfonamide analog and has been shown to inhibit human recombinant caspase 3 and caspase 7 using in vitro models. 18 WC-II-98 was radiolabeled with 18F and evaluated in a rat liver model of apoptosis induced by cycloheximide treatment. 18 Small animal PET images indicate higher uptake of WC-II-98-18F in the liver of rats treated with cycloheximide compared to healthy rats. 18 This suggests that the probe is able to bind to apoptotic cells; however, it does not indicate the sensitivity of this probe since there is a high level of apoptosis induced in the liver model. Further development of this probe requires apoptosis evaluation using a tumor model to determine if the probe is able to detect the lower levels of apoptosis that would be expected after treatment in tumors. Other isatin analogs that inhibit caspase 3 and 7 have been radiolabeled with 18F for development into radiotracers. 20 Recently, a caspase 3/7-specific isatin sulfonamide has been radiolabeled with 18F and evaluated for imaging apoptosis in a murine lymphoma model treated with cyclophosphamide. 21 This tracer was able to detect tumor apoptosis 24 hours after treatment; however, the PET images indicate high retention of the tracer in liver and intestines that may limit its value, especially in the abdominal region. 21 This tracer is likely to be metabolized in the liver and eliminated via hepatobiliary excretion and therefore does not have optimal biodistribution for development into a clinical tracer for imaging apoptosis. Caspase probes have not been tested in humans and this class of probes warrants further development and preclinical testing.
Small-Molecule Probes
The compounds under development are membrane permeable and respond to alterations in the membrane potential and phospholipid scrambling, allowing accumulation inside cells undergoing apoptosis. 22 The compound NST-732 (5-dimethylamino)-1-napththalene-sulfonyl-α-ethyl-fluoroalanine, M W = 368) contains a fluorophore and binds to apoptotic cells early in the biochemical process; however, it is not known where this compound is binding in the cell. 23 NST-732 was tested in three cell lines: human adult T-cell leukemia Jurkat cells, mouse lymphoma Ly-S cells, and CT26 colon adenocarcinoma cells. After treatment of cells with either anti-Fas antibody or the anticancer drug carmustine, NST-732 bound to apoptotic cells. 23 NST-732 was also evaluated in three ex vivo animal studies: radiation-induced cell death in lymphoma; renal ischemia and reperfusion; and cerebral stroke. Uptake of NST-732 was correlated with cell death based on histology analysis for the three animal studies. 23 The authors of this study suggest that since NST-732 contains a fluorine atom it could be radiolabeled with 18F and used as a PET tracer. 23 A different group prepared the compound dansylhydrazone (DFNSH), which binds to cells undergoing apoptosis 24 and subsequently radiolabeled it with 18F. Since this initial publication, there has been no report of testing 18F-DFNSH in cell culture or animal models of apoptosis.
Other low–molecular-weight compounds such as butyl-2-methyl-malonic acid (ML-9) and 2-(5-fluoro-pentyl)-2-methyl-malonic acid (ML-10) are able to bind to cells undergoing apoptosis; however, the biological binding site should be determined if such compounds are to be optimized for clinical use. 25,26 3 H-ML-9 was evaluated by autoradiography using a mouse colon carcinoma model after chemotherapy. 25 3 H-ML-9 indicated increased uptake in tumors after therapy compared to before therapy. 25 This compound could be developed into a radiotracer by radiolabeling with 99mTc or 18F and re-evaluated in an animal model of apoptosis. ML-10 was radiolabeled with 18F and used as a microPET probe to assess neurovascular apoptosis in a mouse model of ischemic cerebral stroke in vivo. 22 18F-ML-10 has proceeded into phase-I/-II clinical trials for imaging of apoptosis. 9
There are two phase-II clinical trials currently recruiting patients for the evaluation of 18F-ML-10 as a PET radiotracer for early detection of response of brain metastases after XRT. 27,28 Recently, it was reported that 18F-ML-10 is able to detect cell death induced by XRT in patients with brain metastases. 29 PET images from 10 patients were obtained before initiation of treatment and on day 9 or 10 after initiation of XRT. 29 Results of this initial study indicate that the signal in tumors increased after irradiation. 29 The use of 18F-ML-10 in the clinic may be feasible given that it has shown some success in phase-I clinical trials, has financial support for future clinical trials, and is able to detect tumor response after XRT. Future research should include evaluation of tumor response to chemotherapy and testing of 18F-ML-10 in other tumor models.
Annexin V-Based Probes for Imaging Apoptosis
Annexin V is a naturally occurring 36 kDa human phosphatidyl binding protein that binds with high affinity to externalized PS on apoptotic cells. 30 Annexin V has been widely investigated as a probe for the detection and molecular imaging of apoptosis. Fluorescently labeled annexin V analogs are frequently used for the assessment of in vitro apoptosis with techniques such as flow cytometry and confocal microscopy. 31 –33 The use of annexin V as a potential radiopharmaceutical has some limitations, including the requirements of (1) its production as an expensive human recombinant protein, and (2) calcium for binding to PS. 34 Despite these challenges, annexin V is the most established probe for imaging apoptosis, and the in vivo and clinical evaluation of this probe is discussed in turn. As a cautionary note, in vitro analysis indicates that radiolabeled annexin V can also bind to necrotic cells; therefore, studies with annexin V need to be initially validated using a second molecular imaging probe to detect and confirm apoptosis. 35
Annexin V for imaging apoptosis in vivo
Radiolabeling annexin V with various radionuclides results in the production of imaging agents for use with single photon emission tomography (SPET) and PET. SPET is an imaging modality that can image noninvasively by making use of radioisotopes (such as 99mTc) that emit γphotons (one photon/radioactive decay) that are detected by a γ camera. The development of annexin V into a SPET tracer has been studied in depth, mostly focusing on the use of 99mTc and 125I labeling. Annexin V was derivatized with hydrazinonicotinamide (HYNIC) and coupled to 99mTc and subsequently evaluated in three animal models of apoptosis: hepatic apoptosis induced by anti-Fas antibody in mice; acute rejection in rats with transplanted cardiac allografts; and cyclophosphamide treatment of transplanted murine B cell lymphomas. 36 Imaging in these animal models indicated a promising two- to sixfold increase in uptake of annexin V at sites of apoptosis. 36 Hepatoma tumors in rats showed significantly increased uptake of 99mTc-HYNIC-annexin V after a single dose of cyclophosphamide compared to untreated rats. 37 Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL), an established assay for apoptosis, was used to analyze tumor tissue samples, and the results indicated that TUNEL-positive cells in the tumor sample correlated well with the uptake of 99mTc-annexin V. 37 The feasibility of using 99mTc-annexin V for repetitive detection of apoptosis was evaluated in a rat model of hepatoma treated with cyclophosphamide, and the results indicated that annexin V can be useful in this context, which is critical for the clinical use of this tracer. 38 Longitudinal monitoring is especially important given that the baseline detection of apoptosis before treatment will be required for correct analysis of apoptotic response to therapy. Annexin V labeled with 125I was evaluated in ependymoblastoma tumor xenografts in mice after irradiation with 2, 5, or 10 Gy of X-rays. 39 Three (3) hours after 125I-annexin V injection the animals were sacrificed, and autoradiographic analysis on tissue slices suggested that annexin V was significantly bound to irradiated tumors. 39 The use of XRT to induce apoptosis in tumor models was recently evaluated in murine models of lymphoma and sarcoma by obtaining SPET images 24 hours after XRT using the tracer 99mTc-HYNIC-annexin V. 40 Tracer uptake in lymphoma increased as the XRT dose escalated from 0 to 8 Gy. 40 Annexin V uptake in lymphomas also correlated with apoptosis from the histology analysis. 40 However, when xenograft sarcoma tumors were treated to 8 Gy, the uptake of 99mTc-HYNIC-annexin V was similar to the background and the tumor was not clearly defined in the images. 40 99mTc-HYNIC-annexin V may therefore be useful in some cases to detect radiation-induced apoptosis in vivo (or lack thereof) and may be predictive of radiation response of different tumors; however, caution would be required given that imaging with 99mTc-HYNIC-annexin V may not be useful for some tumor types, such as sarcoma tumors, and this imaging technique should be validated for specific tumor types. The absence of apoptosis in these sarcoma xenografts after treatment with XRT using the tracer 99mTc-HYNIC-annexin V clearly warrants further investigation to determine which mechanism of cell death occurs in this tumor after XRT. Other tracers may be more useful for this type of tumor, and we discuss tracers for imaging other cell death mechanisms below.
The development of annexin V into a PET tracer is still underway. Keen et al. directly labeled annexin V with 124I and evaluated this compound in a mouse liver model of apoptosis induced by anti-Fas antibody. 41 The PET images indicate that 124I-annexin V localized in apoptotic liver. 41 However, this group did not compare with other tracers such as annexin V labeled with 99mTc or 18F; therefore, it is not clear if 124I imparts any advantages as a radionuclide. The half-life of 124I is 4.2 days, and this and other unfavorable radionuclide properties will cause patients to be exposed to a high dose of radiation if used in the clinic; further, labeling with 124I is laborious, making it unfavorable for imaging purposes. 42 Murakami et al. labeled annexin V with 18F and analyzed biodistribution in healthy rats and in an ischemia model of apoptosis. 43 Annexin V labeled with 99mTc and 18F was compared to determine which radionuclide enhanced the capabilities of the tracer. 43 Results indicate that 18F-annexin V had lower liver, spleen, and kidney uptake than 99mTc-annexin V. 43 An advantage of 18F-annexin V as a radiotracer therefore is better images due to lower background and use of PET for data acquisition, which is quantitative and has superior image resolution to SPET. When 18F-annexin V was evaluated in a liver model of apoptosis in rats induced by treatment with cycloheximide, PET images displayed a three- to ninefold increased uptake of 18F-annexin V in apoptotic liver. 44 The biodistribution of 18F-annexin V in normal rats showed highest uptake in kidneys and bladder. 44 In addition, 18F-annexin V allows the use of PET imaging modalities that, as noted above, produces better images due to the sensitivity of this modality. However, there is limited research published on the use of 18F-annexin V as a PET radiotracer.
Of the eight articles discussed that used animals to test annexin V, the models used were: hepatic apoptosis 36,41,44 ; ischemia 43 ; allograft rejection 36 ; lymphoma treated with cyclophosphamide 36 ; lymphoma and sarcoma treated with XRT 40 ; ependymoblastoma xenograft tumors treated with XRT 39 ; and liver tumor models treated with cyclophosphamide. 37,38 The authors suggest that these models are all providing an apoptosis target and are therefore capable of evaluating annexin V binding to apoptotic cells in vivo. However, some models will invoke large amounts of apoptosis that are readily detected by the probe. For example, the hepatic apoptosis models cause massive amounts of cell death in the liver, which may not reflect the levels of apoptosis that will be present in animal or human tumors after anticancer treatment. There is, therefore, a critical need to be able to detect the apoptotic signal compared to the background and to develop a tracer that will be specific enough to detect lower signal levels. The hepatoma model may be less than ideal given that there is liver uptake of annexin V in healthy animals and therefore a high background exists. One consideration for xenograft tumor models is that the tumor microenvironment is usually different depending on where the tumor is located in the body, and this may influence the tumor uptake of tracer and the corresponding images that are produced. Nonetheless, annexin V has proceeded into clinical trials and has been used to evaluate response to therapy in various tumor models.
Annexin V for imaging apoptosis in clinical trials
Annexin V has to date only been evaluated as a SPET tracer in humans. A phase-I study of the radiotracer 99mTc-annexin V determined the safety and feasibility of this probe for imaging of apoptosis in tumors after the first course of chemotherapy. 45 In this study, annexin V was conjugated to bisthioacetamido-pentanoyl (BTAP) using the N2S2 method for labeling. 46 Fifteen (15) patients with lymphoma, lung cancer, or breast cancer were included in the study and underwent 99mTc-annexin V SPET scans before and within 3 days after the first round of chemotherapy. 45 There was no tracer uptake before therapy; however, after therapy, 7 patients indicated significant 99mTc-annexin V tumor uptake (an example of a patient with 99mTc-annexin V tumor uptake is shown in Fig. 1). 45 These patients had a complete or partial response after the full course of therapy. 45 Of the other 8 patients with no 99mTc-annexin V tumor uptake, 6 had progressive disease. 45 The 2 patients with no 99mTc-annexin V tumor uptake and no progressive disease were patients with breast cancer. 45 The use of 99mTc-annexin V in this small-scale analysis was therefore useful for predicting response of lymphoma and lung cancer but was not predictive of breast-cancer response. Since only 2 patients with breast cancer were included in this study, the effectiveness of 99mTc-annexin V should be evaluated in a clinical trial with a larger cohort of patients with breast cancer. 99mTc-annexin V is safe for clinical use and biodistribution of the tracer includes uptake in salivary glands, liver, spleen, bone marrow, colon, kidneys, and bladder. 45 The BTAP form of annexin V used by Belhocine et al. has a major disadvantage of hepatic uptake and excretion into the digestive system that prevents imaging of the abdomen (Fig. 1B). 45
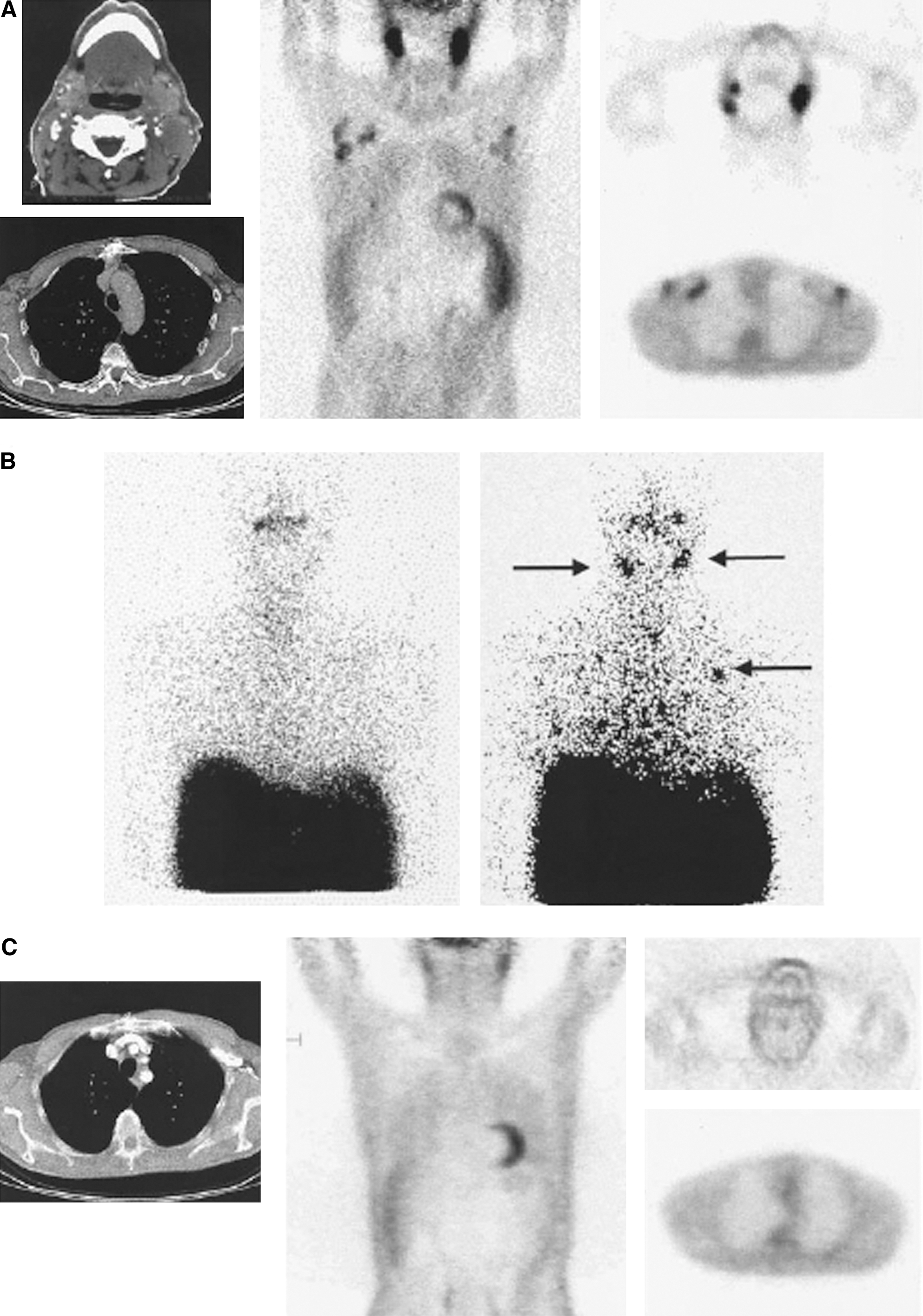
A case of non-Hodgkins lymphoma (stage IV) treated by cyclophosphamide-doxorubicine-vincristine-prednisone protocol with a positive 99mTc-annexin V study.
Contrast level is defined as the signal (apoptosis)-to-background (no apoptosis) ratio. 46 The better the contrast level for a radiotracer, the better the quality of images will be. 99mTc-HYNIC-annexin V accumulates in kidneys and is taken up by liver, red marrow, and the spleen and is excreted in the urine. 42 However, compared to BTAP annexin V form, the HYNIC form has a more favorable biodistribution given that there is less uptake in the intestines and bowel. 46 Labeling of annexin V-HYNIC with 99mTc is well-established in a premade kit 42 and the majority of human studies have used 99mTc-HYNIC-annexin V.
Evaluation of 99mTc-HYNIC-annexin V in healthy male volunteers indicated that it has a better biodistribution than the radiotracer used by Belhocine et al. 45 (BTAP form) given that there is less uptake in the abdomen. 47 This form of radiolabeled annexin V has been studied the most in human patients for the purpose of imaging apoptosis. Unfortunately, 50% of this tracer's injected dose accumulated in the kidneys 3 hours after injection. 47 Other organs that indicated significant accumulation were the liver, red marrow, and spleen. 47 Future apoptosis-indicating probes should be designed to have less uptake in these organs and hence a more favorable biodistribution.
99mTc-HYNIC-annexin V SPET was evaluated in 11 patients with lymphoma after XRT, and this was the first study to image radiation-induced apoptosis in patients with lymphoma at early stages of treatment. 48 Follicular lymphoma was chosen because it is highly sensitive to XRT and undergoes rapid onset of response. 48 Tumor 99mTc-HYNIC-annexin V uptake was increased after two fractions of 2 Gy XRT and the increased uptake correlated with the appearance of apoptotic morphology as determined by cytology and subsequent clinical outcome. 48
99mTc-HYNIC-annexin V SPET was used in another study by Kartachova et al. 49 to evaluate tumor response to therapy in 33 patients with malignant lymphoma, leukemia, non–small-cell lung cancer (NSCLC), and head and neck squamous-cell carcinoma (H&NSCC). XRT was used to treat 27 patients, chemotherapy for 5 patients, and 1 patient received both. Complete or partial response was associated with an increase in annexin V uptake soon after treatment initiation compared to baseline (before treatment). 49 Apoptosis was possible to detect after treatment with either XRT or chemotherapy. The tumors in the study were considered both apoptosis sensitive (lymphoma) and apoptosis resistant (NSCLC and H&NSCC). The results suggest that annexin V may be used as a predictive assay for early treatment response. Unfortunately, there is a need for a larger patient cohort and awareness that annexin V will not discriminate between apoptotic cells versus necrotic cells. 49
NSCLC is a malignancy for which early knowledge of potential response to therapy could significantly help clinicians and improve patient outcomes. Chemotherapy results in a modest improvement in patients with advanced lung cancer; however, chemotherapy is also associated with significant toxicity. 50 Sixteen (16) patients with NSCLC scheduled for platinum-based chemotherapy were evaluated with 99mTc-HYNIC-annexin V SPET before and 48 hours after start of therapy. 50 In this study, SPET scans at 48 hours showing an increase in annexin V tumor uptake correlated with patient response (complete or partial response). 50 Progressive disease was associated with a decrease in annexin V tumor uptake. 50 Compared to small-cell lung cancer, NSCLC is relatively insensitive to chemotherapy. This may be due to the frequent alteration of p53 and development of resistance to apoptosis. In one study of 118 NSCLC specimens, p53 alterations were detected in 63% of the tumors. 51
H&NSCC is treated with a course of chemoradiation therapy that can be effective yet accompanied by toxic side-effects. 52 99mTc-HYNIC-annexin V SPET was performed in 13 patients with H&NSCC before and 48 hours after the start of chemoradiation; however, there was no correlation between annexin V tumor uptake and patient outcome. 52 This may be partly due to baseline necrosis in these tumors (before treatment) that confounds the use of 99mTc-HYNIC-annexin V SPET as a predictive tool. A previous study found that imaging of apoptosis can be quantitative even if using a SPET radiotracer; however, this result is only apparent when there is an absence of necrosis in the tumor. 53 Van de Wiele et al. 53 found that the uptake of 99mTc-HYNIC-annexin V in tumors of patients with head and neck cancer correlated well with the number of apoptotic cells derived from TUNEL assays of the tumor from surgical resection, supporting the validity of the imaging approach.
In 2001, Theseus Imaging Corporation (Cambridge, MA) started phase-II/-III clinical studies using ApomateTM (99mTc-HYNIC-annexin V) for the detection of early response to chemotherapy in patients with NSCLC. 54 Despite some success in clinical trials with Apomate, Theseus Imaging Corporation unit was closed by North American Scientific in September 2004. 55 Currently, there is a lack of Good Manufacturing Practice (GMP)–grade 99mTc-HYNIC-annexin V kits for clinical imaging trials, and there is a need to complete studies of patients with NSCLC and non-Hodgkins lymphoma; however, the studies may not be completed due to the closure of Theseus. 56
There is some concern about the optimal time for imaging cell death after treatment of breast cancer. 57 Baseline uptake of 99mTc-annexin V has been identified in some tumors. 57 This highlights the requirement to obtain baseline images to determine whether treatment is in fact inducing new cell death. 57 Experience with 99mTc-annexin V indicates that the optimal time for imaging apoptosis due to chemotherapy treatment is 24–72 hours after the first dose of chemotherapy. 46 Timing is also important because apoptotic cells will be removed by macrophages and the rate of apoptosis may be specific to tumor type.
Due to the variation of kinetics of cell death, the optimal imaging time for acquiring images with annexin V may vary with the type of therapy and possibly also with tumor type. 46 It remains unclear if multiple annexin V scans will improve assessment of response. 46 Conventional SPET imaging may miss some of the apoptotic cells within a tumor due to poor contrast level. 46 99mTc-annexin V studies with human patients have shown that it can sometimes be a useful predictive diagnostic assay for treatment response early in therapy. 58 Despite the development of annexin V into a SPET tracer, it is not particularly promising because the chemistry possibilities for enhancing this tracer are almost exhausted and because the funding for the commercialization of this probe is lacking. Future research should focus on the development of annexin V into a PET tracer to obtain better quality images.
PS-Binding Peptides
PS-binding peptides may provide advantages as imaging agents because they are smaller than the PS-binding annexin V and this may contribute to a more favorable biodistribution for imaging purposes. Recently, research has been published by various groups using phage display to isolate peptides that bind to PS and cells undergoing apoptosis. 59 –62 These peptides are candidates to be radiolabeled with radionuclides and evaluated as radiotracers to image apoptosis. A PS-binding peptide isolated by Laumonier et al. has been evaluated in PS-binding assays but has not yet been evaluated in cell models of apoptosis. 59 A fluorescently labeled peptide isolated by Hong et al. was shown to bind to ischemic tissue from a rat model without binding healthy tissues. 61 Thapa et al. evaluated a PS-binding peptide in H460 and H157 human lung cancer cells and U937 leukemia cells undergoing apoptosis after etoposide treatment. 60 An optical imaging technique was also used ex vivo to evaluate the fluorescently labeled peptide in tumor-bearing mice (H460 lung cancer xenograft model) treated with a single dose of camptothecin. 60 The results of optical imaging indicate that the peptide specifically bound to apoptotic cells and surprisingly to tumor vasculature. 60 Vascular endothelial cells in tumors, but not in normal vasculature, externalize PS, which may be caused by oxidative stress and activating cytokines associated with the tumor. 63 PS externalization in tumor vasculature would explain the optical imaging results using a fluorescently labeled peptide; however, this finding reinforces the importance of imaging before and after treatment to analyze changes in PS externalization due to treatment of the tumor.
Recently, three PS-binding peptides were isolated by phage display technology and shown to bind to apoptotic cells with high affinity. 62 One selective and high-affinity peptide (K d* = 2 × 10−9 M) was selected for development into an MRI contrast agent by conjugating to diethylenetriaminepentaaceticacid–isothiocyanate and complexing with gadolinium chloride. 62 This contrast agent was tested with apoptotic Jurkat cells treated with camptothecin and in a mouse model of liver apoptosis in wild-type (WT) mice induced by anti-Fas antibody. 62 This research group is interested in the contribution of apoptosis to the instability of atherosclerotic lesions and aimed to image apoptosis in plaques to diagnose plaques vulnerable to rupture. 62 Apoptosis of macrophages and smooth-muscle cells contributes to the expansion of the necrotic core and thinning of the fibrous cap, which could possibly play a role in plaque rupture. 62 Atherosclerotic lesions were imaged with MRI in ApoE−/− transgenic mice, indicating that this agent could potentially be used to image apoptosis in various disease states. 62 However, a major limitation of this MRI contrast agent is the low sensitivity, which limits detection of apoptosis in regions with low PS concentration. 62 This challenge might be overcome by preparing radiolabeled analogs of the peptides and by using SPET or PET imaging techniques.
Various PS-binding peptides have shown high-affinity binding to apoptotic cells, suggesting that these peptides should be radiolabeled with various radionuclides for development into novel PET and SPET probes. Currently, only one study evaluated a PS-binding peptide for molecular imaging purposes using autoradiography. The peptide from Hong et al. was radiolabeled with 131I, and images indicate that the peptide bound to ischemic lesions in a rat model of apoptosis. 61 Labeling peptides with 18F would allow the development of novel probes for PET. PS-binding peptides have not been tested in humans and require further development and pre-clinical evaluation.
Imaging Premature Senescence
Premature senescence (also referred to as accelerated senescence or stress-induced premature senescence) is a genetically programmed response to DNA damage that is regulated by the p21WAF1 protein (hereafter simply called p21). 64,65 p21 is encoded by the p21 WAF gene that is transcriptionally transactivated by the p53 tumor-suppressor protein in response to genotoxic stress caused by DNA-damaging agents such as ionizing radiation and various anticancer chemotherapeutic drugs. Premature senescence was originally described in fibroblasts, where exposure to ionizing radiation was seen to trigger a permanently growth-arrested phenotype with a striking resemblance to telomere-based replicative senescence. 66,67 This mode of cell death has also been observed in many human tumor cell lines that express WT p53 after exposure to ionizing radiation and chemotherapeutic drugs. 68 –70 Irradiation of some p53-deficient tumor cells also appears to activate at least some aspects of the premature senescence program, although to a much lesser extent than in p53-WT cells (R. Mirzayans and D. Murray, unpublished).
Current in vitro markers for cells undergoing premature senescence in response to genotoxic stress include (1) marked alterations in cell morphology, including enlargement and flattening, although the cells do retain cell membrane integrity (viability) and continue to function metabolically; (2) acquisition of senescence-associated (-galactosidase activity 69 –71 ; and (3) sustained upregulation of the p21 gene/p21 protein in the nucleus. 66,70,72 –76 The role of p21 in activating premature senescence appears to involve multiple mechanisms, including the inhibition of various cyclin-dependent kinases, thereby preventing cell-cycle progression, 77 suppression of DNA synthesis via an interaction with proliferating cell nuclear antigen, 77 and modulation of gene expression. 78 –80 The loss of cellular clonogenic potential associated with premature senescence may be an important contributor to tumor control after both chemotherapy 81 and XRT. 82
Potential for imaging p21 transcripts by PET using 18F-labeled antisense oligonucleotide probes
Currently, there are no validated probes for the in vivo imaging of senescence. Although senescence-associated (-galactosidase staining has been performed with murine tumors treated with cyclophosphamide in vivo, this necessitated invasive sectioning of cryopreserved tissue. 81 One approach that could prove useful for the noninvasive in vivo monitoring of premature senescence in tumor cells post-therapy using PET involves imaging p21 gene expression using 18F-labeled antisense oligonucleotide probes (asODNs). 83 asODNs are small synthetic molecules that can bind to their complementary target mRNA in cells, and thus they have the potential for use as probes for the in vivo molecular imaging of specific gene transcripts. 84 –86 Given the abovementioned key role of sustained p21 gene activation in driving the premature senescence response to DNA damage in vitro, the ability to noninvasively monitor changes in the levels of this transcript in situ could provide an important marker for premature senescence in vivo. An elegant proof-of-principle study for this approach used a breast cancer xenograft model in which tumor p21 mRNA levels were stimulated using epidermal growth factor; the tumors were successfully imaged with a gamma camera using 111In-labeled anti-p21 asODNs. 85 As noted in previous sections of this review, the extension of this technology to PET imaging should provide a significant increase in sensitivity and utility and is currently being evaluated by our group. On the basis of previously described sequences, 85,87 we have synthesized a fully phosphorothioated 18-nucleotide asODN targeted to the 3'-untranslated region of the human p21 mRNA. We have validated this asODN for targeting p21 mRNA in vitro in HCT116 human colon cancer cells 88,89 and have seen no loss of activity after radiofluorination (with 18F) of these ODNs using an established protocol. 90 To date, this probe has not been tested in in vivo models, but it is a promising approach that warrants further investigation.
Other approaches
It may be possible to use standard PET imaging techniques to monitor cells undergoing premature senescence in vivo by focusing on the unique biology of these cells. Proliferating tumor cells are typically positive for 18F-FDG, which, as noted in section 2, is believed to reflect their increased glycolytic rate and glucose avidity, 91 and they are also positive for 3'-deoxy-3'-[18F]-fluorothymidine (18F-FLT), which measures cell proliferation independent of cellular metabolism. 92 As noted above, a hallmark of tumor cells that have undergone premature senescence is that they maintain a significant level of metabolic activity despite being permanently growth arrested. 64 It is therefore possible that whereas proliferating tumor cells will be both FDG+ and FLT+, those tumor cells that activate the premature senescence program after therapy will cease to proliferate (and thus exhibit minimal FLT uptake) but will maintain significant metabolic activity (and thus will continue to take up FDG).
Although this hypothesis has not been tested directly, there are anecdotal examples in the literature in which tumors were irradiated and monitored at early times using both FDG and FLT. For example, Yang et al. exposed SCCVI murine squamous cell carcinoma tumors to X-rays and monitored their response using PET imaging; these tumors were initially 18F-FDG+/18F-FLT+ by PET imaging, but became dramatically 18F-FLT− at 24 and 48 hours after a single exposure to 10 or 20 Gy of X-rays while retaining full 18F-FDG-avidity (Fig. 2). 93 The authors suggested that this effect might be caused by an anticipated suppression of the enzyme thymidine kinase by X-rays 93 ; although this may indeed be the explanation in whole or in part, this is also the behavior that might be expected of a tumor in which the premature senescence phenotype was playing a significant role after XRT. The creative use of 18F-FDG and other recently introduced clinical PET tracers such as 18F-FLT may have some value in determining the specific tissue response to therapy. However, these measurements will always be nonspecific and therefore do not directly measure cell death.
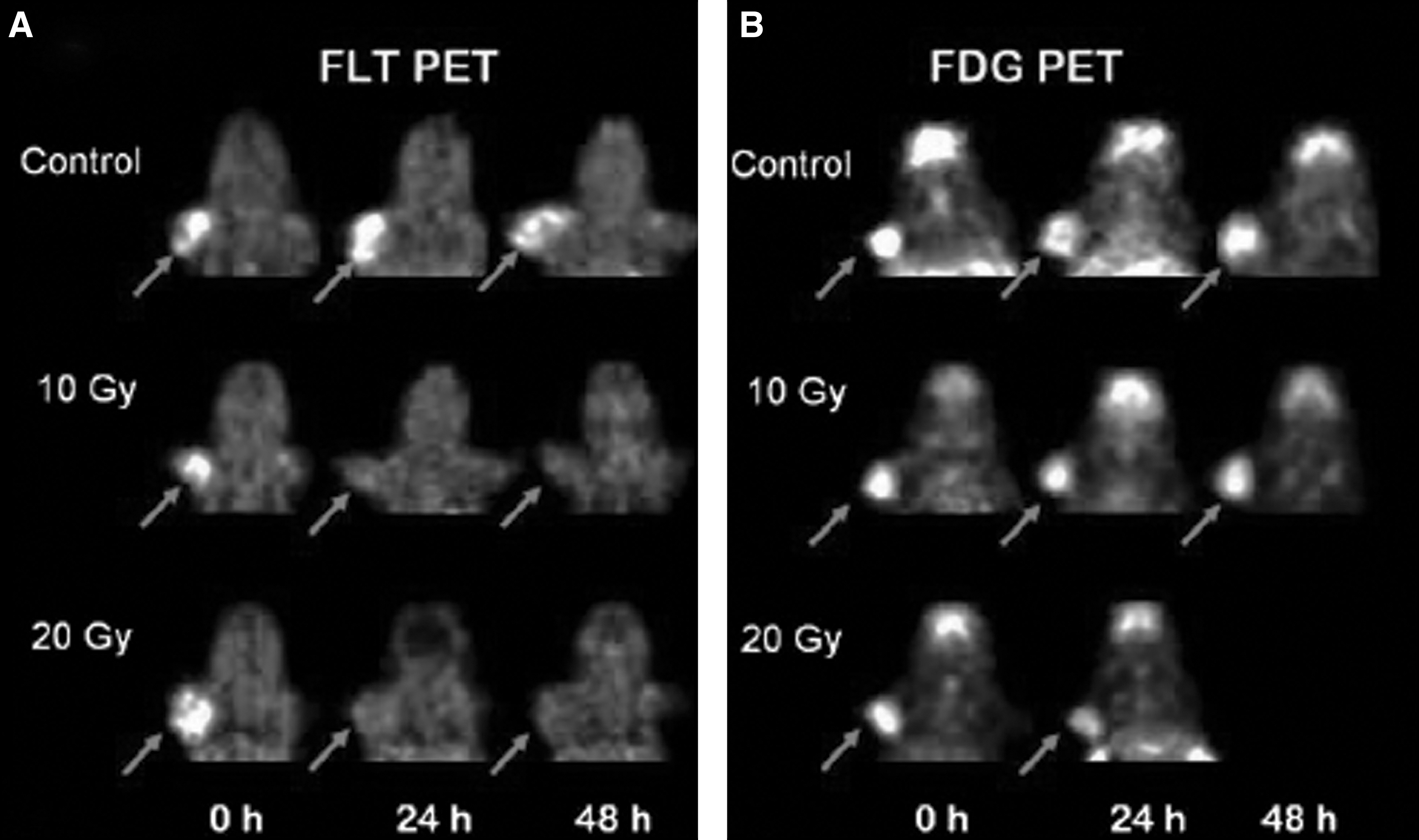
Serial PET images of murine small cell carcinoma VII tumors after radiation treatment.
Imaging Necrosis
Necrosis is generally regarded as a nonspecific (i.e., not genetically regulated) mode of cell death characterized by cell swelling, denaturation/coagulation of cytoplasmic proteins, random fragmentation of DNA, and disintegration of subcellular organelles and of the cell membrane with release of cytotoxic cell components. 5 Necrosis is usually determined in vitro using microscopy analysis. 58 There has been some development of necrosis probes based on the synaptic vesicle membrane protein synaptotagmin. This protein does not specifically bind to PS; however, it does bind to anionic phospholipids (such as PS and phophatidylethanolamine) that are exposed when the cell membrane is disrupted and that might be used to indicate necrosis. 94,95 The C2A domain of synaptotagmin has been radiolabeled with 99mTc and used as a molecular probe for noninvasive imaging of acute myocardial infarction and ischemia. 95 –97 Synaptotagmin-99mTc has been evaluated in a healthy rat model and in a rat model of acute myocardial infarction. 95 Myocardial infarct and ischemia is clearly visible in the SPET images obtained with this tracer. 95 Synaptotagmin-99mTc has been evaluated in a mouse model of NSCLC and found to bind to tumors after chemotherapy; however, this tracer also indicated uptake in kidneys and liver. 98 Synaptotagmin has not been evaluated in humans, and its clinical use is uncertain. It may have some applications that warrant use in clinical practice; however, due to its lack of specificity it may not be as powerful as other tracers under development. Other probes for imaging necrosis take advantage of the characteristic loss of cell membrane integrity, and these have been recently reviewed. 9 Other compounds that can bind to cells with loss of membrane potential could potentially be developed into radiotracers. Future work should focus on developing new potential probes for in vivo imaging of necrosis to generate a specific probe for necrosis (i.e., that do not overlap with PS/apoptosis) that will help to differentiate cell death response(s) after treatment.
Imaging Autophagy
Autophagy is a conserved stress response in which cells exit the cell cycle, autodigest proteins and damaged organelles, shrink, and recycle amino acids and fatty acids. 99 It is controlled by pathways that signal through the mammalian target of rapamycin protein and occurs in some tumor cell lines after exposure to XRT or chemotherapeutic agents. 100,101 It is unclear if autophagy provides cancer cells with an escape mechanism or if autophagy is used to eliminate tumor cells in the body. 3 It is also not clear whether autophagy is a separate form of cell death or if failure of autophagy to rescue cells from stress can lead to cell death by either apoptosis or necrosis. Currently, transmission electron microscopy is the standard method for in vitro detection of autophagy. 58 Microtubule-associated protein light chain 3 (LC3) is now widely used for in vitro detection of autophagy. 102 Endogenous LC3 is detected as two bands after sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotting. One band is LC3-I, which is cytosolic, and the other is LC3-II, which is bound to phosphatidylethanolamine and is present on autophagosomes. 102 The amount of LC3-II is correlated with the number of autophagosomes and experiments can detect the LC3 conversion of LC3-I to LC3-II. 102 The development of a radiotracer specific for autophagy is a possibility if a compound could be isolated that binds specifically to LC3-II with high affinity. Autophagy may be an important mode of tumor cell death after treatment, but, at present, we do not have a specific method to image it in vivo. 103 A basic research initiative is required to, first, identify a target that is unique to autophagy and, second, to develop an imaging probe based on this target.
Summary and Future Directions
Our present understanding of cell death is mainly derived from observations on cells in vitro and ex vivo tissues. There is now an increasingly clear picture of the distinct modes of cell death in terms of their morphological changes and the genomic and proteomic changes that are characteristic of each mode. This remains an area of vigorous research activity including applying the observations made in cells and isolated tissues to the investigation of cell death in vivo. In this context, and as outlined in this review, we are starting to see the development and preclinical and clinical exploitation of radionuclide-based molecular imaging probes with the capacity to selectively and noninvasively image the different modes of cell death.
Apoptosis has been the most intensively studied mode of cell death and the majority of tracer development has focused on this pathway. The four classes of imaging agents for apoptosis described in this review are (1) caspase binding probes in preclinical development, (2) low-molecular-weight probes in phase-II clinical trials for PET imaging, (3) annexin V in phase-II/III clinical trials for SPET imaging, and (4) PS-binding peptides in pre-clinical development. Imaging of premature senescence is being approached using 18F-labeled asODN probes to image p21 transcripts with PET. Necrosis has one main tracer, synaptotagmin, in preclinical development as a SPET radiopharmaceutical. The role of autophagy in tumor response to therapy is not completely understood; however, the development of probes to image autophagy would help to explore the prevalence and define the importance of this pathway.
A number of the radiotracers in use or under development for imaging cell death are analogs of targeting agents first applied as fluorescent probes for in vitro cell studies or for probing tissues from animal studies or human biopsy. This is the case for annexin V, where the radionuclide probes labeled with 99mTc or 18F have exploited the known PS targeting of fluorescent probes such as annexin V fluorescein isothiocyanate.
104
Thus, other fluorophore-based probes should be considered as lead compounds for potential radionuclide probes. For example, a recently developed fluorogenic substrate, 5-dodecanoylaminofluorescein di-ß-
In spite of recent developments in identifying targets and probes for cell death and the progression of several of the radiolabeled probes into preclinical and clinical development, there are a number of significant challenges to effectively imaging the various modes of cell death in vivo. Imaging needs to be performed before and after treatment and the protocol needs to be consistent in how the images are obtained. Other factors to consider include the injection dose, type of SPET or PET camera used to obtain images, acquisition protocol, software for analysis, and interpretation criteria. 46 These factors need to be consistent in multicenter clinical trial evaluation of radiotracers and the same factors also need to be consistent in patients when obtaining pre- and post-therapy images. As noted above, tumor cells may also die at varying rates, and the time course also depends on the type of treatment. This is important to consider when evaluating a radiotracer in various cancers and associated therapies. In vivo, this may be due to many factors, including the tumor microenvironment and accessibility of the tumor to vasculature. Thus, any effective imaging protocol will need to explore these features to provide appropriate guidelines for the timing of imaging after treatment.
Conclusions
The overall message of this review is that early tumor response to therapy is very complex. This review discusses four possible cell death mechanisms, each of which represents unique and complex pathways. The use of radiotracers to image individual cell death mechanisms would provide valuable information on tumor response in a rapid and noninvasive manner, and this information could be used to make subsequent decisions about patient treatment. Ideally, these imaging agents would be specific for one mode of cell death and would be properly validated and capable for use with PET. Currently, the mechanism of cell death is unknown for most solid tumors, and tumor regression may actually be due to a combination of cell death mechanisms resulting in overall tumor size reduction. 5,81 Responses at the cellular level can have profound clinical implications in selecting and applying effective therapy. Given that tumor cells can lose their clonogenic potential through several different pathways, it will be critical to better understand and monitor modes of cell death in tumors from individual patients to better exploit interindividual differences when combining conventional genotoxic therapeutics with modifying agents. Thus, there would appear to be a clinical role for imaging agents that could identify the specific cellular response to therapy. Imaging agents would also have an important role to play in preclinical investigations in animal models in helping to understand the induction and progression of the various cell death mechanisms and in drug development studies. Although this review has concentrated on cancer, it must be remembered that cell death is a factor in many diseases both as an acute event (stroke and heart attack) or chronic progression (arthritis and neurodegeneration). Consequently, we can expect that the research efforts will continue as we attempt to fully understand the cell death mechanisms and to develop probes for in vitro and in vivo imaging.
Footnotes
Acknowledgments
J.K. is supported by Alberta Cancer Research Institute Graduate studentship and Killam scholarship.
Disclosure Statement
No conflict of interest exists for any of the authors.