
Abstract
Although hypoxia can induce cell death, the cancer cells and endothelial cells within a solid tumor that remain active in the hypoxia microenvironment often possess an enhanced survival potential. Developing approaches aimed at increasing the sensitivity of endothelial cells to hypoxia-induced cell death represents a potentially important avenue for antiangiogenesis treatment. This study investigated approaches to increase the sensitivity of endothelial cells to hypoxia-induced apoptosis. Autophagy and apoptosis of endothelial cells induced by hypoxia were investigated by transmission electron microscopy, confocallaser microscopy, and western blotting. Moreover, cell invasion was observed by a transwell assay and F-actin quantitative analysis. In this study, it was found that hypoxia could induce both autophagy and apoptosis in hypoxia-inducible factor-1- and Beclin1-dependent endothelial cells. Hypoxia-induced autophagy was prohibited by phosphatidylinositol 3-kinase/Akt inhibitor but not mitogen-activated protein kinase inhibitor. Inhibition of autophagy promoted the rate of apoptosis. Further, the reversal of hypoxia-induced autophagy increased cell migration compared with the normoxia condition. This study concludes that hypoxia triggers a feedback mechanism that delays apoptosis of endothelial cells and that is driven by hypoxia-induced autophagy. Thus, approaches aimed at the disruption of this mechanism can be expected to enhance the susceptibility of endothelial cells to hypoxia-induced apoptosis.
Introduction
Low oxygen tension, a pathophysiological condition of solid tumors that exacerbate cancer prognosis, is associated with resistance to radiotherapy and chemotherapy due to deficient or aberrant blood flow. 1 Several groups have shown that, in general, tumor cells are well adapted to moderately low-oxygen environments. 2,3 However, as extreme hypoxia is capable of activating apoptosis through a hypoxia inducible factor-1 (HIF-1)-dependent pathway, the consequence of tumor hypoxia can also be other types of programmed cell death such as autophagy. 4 Moreover, recent studies have suggested that hypoxia could induce autophagy in a model of the ischemia/reperfusion condition. 5
Autophagy is an intracellular, proteolytic event that plays an important role in the turnover of long-lived intracellular proteins and organelles. Indeed, autophagy principally serves an adaptive role to protect cells against diverse pathologies, including infections, hypoxia, defective nutrition, and other stress conditions. 6 The double-membrane structures called autophagosomes or autophagic vacuoles fuse with lysosomes and mature into autolysosomes or autophagolysosomes, where their contents are degraded by acidic lysosomal hydrolyses. 7 The process may serve to regulate normal turnover of organelles and to recycle those with compromised function to maintain homeostasis. In addition, research has shown that angiogenesis inhibitors such as Kringle 5 and Endostatin can induce autophagy, which is a potential target for antiangiogenesis cancer therapy. 8,9 Beclin1, the mammalian homologous gene of the yeast Apg6/Vps30 gene, a major mediator of autophagy, can stimulate autophagy when overexpressed in mammalian cells. 10 Beclin1 can also bind to Bcl-2, an important regulator of apoptosis. 11 The relevance of Beclin1 as an autophagic gene has been shown by both experimental studies and human cancer observations. Heterozygous disruption/mutation of the Beclin1 gene causes increased cell proliferation and development of cancer, leading to the conclusion that Beclin1 is an anti-oncogene. Beclin1 is a multimeric complex with class 3 phosphatidylinositol 3-kinase (PI3K), and it is necessary for the formation of autophagosome during the autophagic sequestration process. The complex of class 3 PI3K-Beclin1 was found to be relevant in the regulation of autophagy, and PI3K was recognized as the major physiological partner of Beclin1 in a human glioblastoma cell line. Mitogen-activated protein kinases (MAPKs), particularly MAPKp38, are implicated in amino acid signaling, which regulates autophagy in response to various stimuli. However, the molecular mechanisms of MAPKp38 regulation of autophagy remain widely unknown. Recent studies suggest 12 that MAPKp38 negatively regulates the interaction of Atg9 with a novel Atg9 binding partner, p38IP, to control the levels of autophagy induced in response to stress.
In a previous study, endothelial cells were found to be the key cell of angiogenesis. 13 However, there are very few studies that evaluated autophagy in endothelial cells under the hypoxic condition. In the present study, it was hypothesized that induction of autophagy in endothelial cells represents a critical adaptive mechanism under hypoxic conditions via the Beclin1/PI3K pathway. To test this hypothesis, an in vitro model was employed to mimic the hypoxia pathological environment, to investigate which hypoxia condition was sufficient to induce autophagy in endothelial cell, to determine whether this response was HIF-1-dependent, and to investigate whether autophagy could be directly and efficiently inhibited by PI3K and MAPK p38 inhibitors.
Materials and Methods
Agent
The anti-MAP1-LC3-II polyclonal rabbit antibody and anti-Beclin1 monoclonal mouse antibody were obtained from Abgent Biotechnology; the GAPDH, HIF-1α, Akt, phospho-Akt, MAPK p38, and phospho-MAPK p38 antibody were obtained from Santa Cruz Biotechnology. HRP-labeled goat anti-rabbit and GFP-labeled goat anti-rabbit immunoglobulin were purchased from Boster. The ECL western blotting detection system was from Millipore. LY-294002, 3-methyladenine (3-MA), SB-203580, and the Caspase-3 activity quantification assay were obtained from Sigma.
The shRNA constructs, targeting Beclin1, were sequences of two different targeted sites in mouse Beclin1: TGGACA CGAGCTTCAAGAT and TGGAGATCCTAGAGC-AGAT. The annealed inserts were cloned into the RNAi-Ready pSIREN-RetroQ vector, which had been previously digested with EcoRI-BamHI.
LC3-GFP construct was kindly provided by Marja (Apoptosis Department and Centre for Genotoxic Stress Research, Copenhagen, Denmark).
Cell culture and cell treatment
The human endothelial cells EAhy926 were obtained from the Institute of Pathology of the Tongji Hospital. They were maintained in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2.
Hypoxic conditions were induced in a sealed Bug-Box anaerobic workstation (Ruskinn Technology Biotrace International). The oxygen level was maintained at 1%, with the residual gas mixture being 94% nitrogen and 5% carbon dioxide.
Cells in the exponential phase of growth were plated in six-well plates and incubated in the anaerobic workstation for 6 and 24 hours in the presence or absence of 2 mM 3-MA, 10 mM LY294002, or 10 mM SB-203580.
Western blot analysis
After treatment, the cells were washed with cold PBS and homogenized in homogenization buffer containing protease inhibitor cocktail (1:1000) in 50 mM Tris (pH 7.0), 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride. The raw homogenate was centrifuged at 4°C for 20 minutes at 12,000 g. The supernatants (20 μg) with 5× loading buffer were heated for 10 minutes at 100°C and separated by 15% SDS-PAGE, after which the proteins were transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% nonfat skim milk in 1× Tris-buffered saline with 0.1% Tween 20 (TBST) for 1 hour at room temperature and then incubated overnight with primary antibodies (1:300) at 4°C. The following day, the blots were washed with TBST (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, and 0.1% Tween-20) for 10 minutes three times and exposed for 60 minutes at room temperature to an appropriate HRP-linked secondary antibody (1:1000). Complexes of the primary and secondary antibodies were visualized using a chemiluminescent substrate kit and exposed to medical X-ray film. Films were obtained and analyzed using ImageJ software.
Electron microscopy
After treatment, the cells were fixed with glutaraldehyde and osmium tetroxide, treated with uranyl acetate lead citrate for enhanced protein and lipid staining, and then dehydrated in ethanol. The cell pellets were embedded in epon resin and cut with an ultramicrotome to 70 nm thickness before viewing. Samples were analyzed in a Tecnai 12 Biotwin (FEI) microscope.
Visualization of monodansylcadaverine vacuoles
The treated cells were labeled with a 50 mM concentration of the autofluorescent marker monodansylcadaverine (MDC) from Sigma in PBS at 37°C for 30 minutes. The cells were then fixed with 3.7% formaldehyde for 10 minutes at room temperature and washed in PBS for 10 minutes three times. The cells were observed by fluorescence microscopy with an Olympus Eclipse.
Immunocytochemistry and confocal microscopy
The cells transfected with the LC3-GFP construct were plated onto polylysine-coated coverslips and then incubated for 24 hours under hypoxia condition. DAPI (Vector Laboratories, Inc.) was used to visualize mitochondria and the nucleus. The cells were first observed with a fluorescence microscope (Olympus) to localize the expression of the green fluorescent protein, followed by a z-stack using a confocal laser microscope (FV500; Olympus) at 300× magnification. Fields were chosen randomly from various sections to ensure objectivity of sampling. Digital images were processed to determine the number of autophagic vesicles per cell.
Flow cytometry
The cells were washed in PBS and resuspended in binding buffer (10 mM HEPES/NaOH [pH 7.4], 140 mM NaCl, and 2.5 mM CaCl2). Annexin V-FITC was added to a final concentration of 100 ng/mL, and the cells were incubated in the dark for 10 minutes, then washed again in PBS, and resuspended in 300 mL of binding buffer. Ten (10) milliliters of propidium iodide (PI) was added to each sample before flow cytometric analysis. The cells were analyzed using a Becton Dickinson FACStar plus flow cytometer (Becton Dickinson). Electronic compensation was used to eliminate bleed-through fluorescence. In each sample, a minimum of 10,000 cells were counted and stored in list mode. Data analysis was performed with standard Cell Quest software (Becton Dickinson).
Caspase-3 activity quantification assay
Caspase-3 activity was assessed using the caspase-3 Colorimetric Assay Kit (Sigma Aldrich), according to the manufacturer's instructions. This assay is based on the detection of the amount of Ac-DEVD-p-NA substrate cleaved by cell lysates to release the colored p-NA molecule. Following treatment, the cells were washed in PBS and suspended in a lysis buffer (50 mM HEPES [pH 7.4], 5 mM CHAPS, and 5 mM DTT) for 15 minutes at a concentration of 107 cells per 100 μL of buffer. Lysed cells were centrifuged at 16,000 g for 15 minutes. Lysate protein concentrations were determined using the Bradford assay. Equal amounts of protein (∼20 μg) from each sample were added to wells containing the assay buffer (20 mM HEPES [pH 7.4], 0.1% CHAPS, 5 mM DTT, and 2 mM EDTA), followed by 10 μL of Ac-DEVD-p-NA (20 mM), bringing the total volume of each well to 100 μL. Caspase-3 activity was assessed by measuring the optical density at 405 nm using a spectrophotometer from Molecular Devices. All experiments were performed in duplicate and repeated at least twice.
Transwell assay
The transwell assay was performed using a 24-well microchemotaxis chamber (Costar). One hundred thousand EAhy926 cells in 200 μL of medium were seeded into the upper chamber (pore size: 8 μm). Then, the cells were treated as previously described. The apparatus was incubated for 16 hours at 37°C in a humidified chamber with 5% CO2 to allow cell migration. After the incubation period, the filter was removed, and the cells on the upper side of the filter that had not migrated were scraped away with a cotton applicator. The filter was fixed for 30 minutes at room temperature with 4% paraformaldehyde, washed twice with PBS, and stained with crystal violet. The number of cells that had migrated through to the lower surface of each membrane was counted. Each experimental point was repeated six times.
F-actin quantification
To visualize F-actin, the cells were stained with FITC–phalloidin using a standard protocol and imaged using a 120× objective. For quantification of fluorescence, images of phalloidin-stained cells from 6 to 12 random fields of observation were acquired in each experiment using a 60× objective. After performing background subtraction of the images, the average fluorescence intensity per cell was calculated for each field of observation.
Statistical analysis
Experiments were done independently three times. Comparisons were made between hypoxia-treated cells and the control group. A two-tailed Student's t-test or one-way ANOVA (SPSS software version 11.5) was used for analysis. p<0.05 was considered to indicate statistical significance.
Results
Hypoxia induces autophagy in a time-dependent manner in endothelial cells
Transmission electron microscopy was used to confirm the nature of hypoxia-induced vesicles (Fig. 1A). An increased accumulation of large vesicles within the cytoplasm of treated cells was observed after a 24-hour treatment, compared with the accumulation observed at earlier time points (0 and 6 hours). A few single membrane vesicles were observed at the edge of control cells. The presence of these vesicles in the control treatment can be attributed to serum restriction.

Hypoxia induces autophagy in endothelial cells.

Endothelial cells were transfected with LC3-GFP, a biomarker for autophagy, and exposed to hypoxia condition for 6 or 24 hours. LC3, a microtubule-associated protein light chain 3 (MAP-LC3), typically exhibits diffuse cytosolic distribution. Representative confocal images, shown in the lower panel (Fig. 1B), confirm that the treatment of hypoxia led to the redistribution of modified LC3 to punctate structures and increased the number of LC3-GFP-positive vesicles in the hypoxia-treated cells. The number of autophagic vesicles per cell increased as the induced time become longer. The mean number of LC3-GFP-positive vesicles in the control endothelial cells was 6±2. After 24 hours of hypoxia conditions, the LC3-GFP-positive vesicles increased to 30±10, which is significantly different (p<0.05) (Fig. 1C).
Autophagosome formation is also known to correlate with the conversion of LC3-I to LC3-II, which involves proteolytic cleavage and lipidation. When autophagy is activated, such as after amino acid starvation, the LC3-I protein localized in the cytoplasm is cleaved, lipidated, and inserted as LC3-II into autophagosome membranes. Thus, an increase in the amount of the smaller-molecular-weight LC3-II protein and an increase in the LC3-II/LC3-I ratio are hallmark of autophagy and correlate with an increased number of autophagosomes (Fig. 1D).
Hypoxia-induced autophagy is Beclin1/PI3K dependent and MAPK independent
The class III PI3K/Beclin1 complex is essential for the vesicle nucleation step in autophagy. To evaluate whether autophagic induction by hypoxia can be inhibited by interfering with the activity of PI3K, LY-294002, a widely used PI3K inhibitor, was added to LC3-GFP-transfected EAhy926 cultures at a nontoxic concentration (Fig. 1B). In addition, reducing the expression of Beclin1 with shRNA also resulted in reduction of the autophagic response. Figure 1C shows that the LC3-GFP-positive vesicles decreased to 12±5 when combining hypoxia with LY-294002 (p<0.05), about 40% of that of the control, and to 14±6 when combined with Beclin1 shRNA. Moreover, to evaluate the effect on the MAPK pathway of using SB-203580, 22±7-positive vesicles were observed in endothelial cells, which decreased by 10% (p>0.05) to the control cells. These data suggest that Beclin1 and PI3K complex but not MAPK pathway play important roles in hypoxia-induced autophagy of endothelial cells.
Hypoxia upregulates Beclin1 levels
A Western blot was used to determine whether hypoxia-induced autophagy in endothelial cells involved Beclin1 and HIF-1 expression (Fig. 2A, B). The expression of HIF-1 and Beclin1 was observed after hypoxia treatment for 24 hours, which was constant when combined with LY-294002 or SB-203580. Levels of Bcl-2 and the Beclin1 achieved a steady state after hypoxia treatment. These data suggest that hypoxia induces autophagy in endothelial cells by upregulating Beclin1 expression.
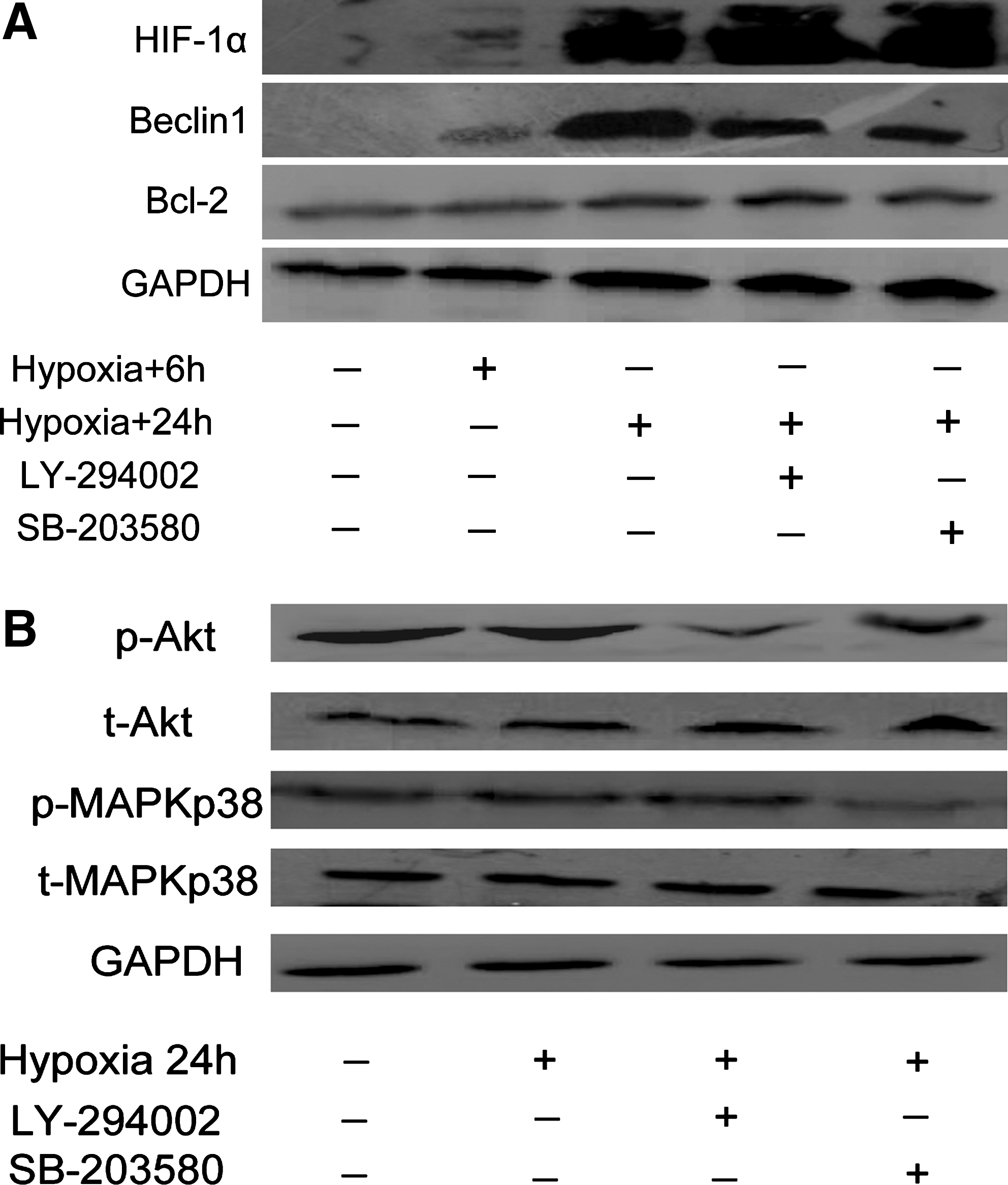
Hypoxia induced changes in Beclin1-Bcl-2/Bcl-xL association.
Inhibition of autophagy in endothelial cells potentiates the apoptotic effect of hypoxia
Hypoxia treatment induced the upregulation of Beclin1 in endothelial cells and simultaneously impinged on the availability of antiapoptotic proteins Bcl-2 and Bcl-xL to bind the protein. This prompted to investigate whether the proapoptotic effect of hypoxia could be enhanced by inhibiting autophagy. Figure 3A summarizes the flow cytometric analysis of Annexin V-FITC/PI activation in EAhy926. Hypoxia treatment of cells with 3-MA showed an increase in Annexin V-FITC/PI-positive cells to 15%, compared with 8% for the control (p<0.05). Moreover, the caspase-3 activity quantification test showed that 6 and 24 hours of hypoxia could increase caspase-3 activity by 235.6% and 288.8%, respectively, compared with the control group. Also, 24 hours of hypoxia combined with 3-MA could increase the caspase-3 activity to 587.6% (p<0.05) (Fig 3B). Together, these data suggest that the inhibition of hypoxia-induced autophagy could increase the apoptosis rate in endothelial cells.

3-Methyladenine (3-MA) upregulates apoptosis induced by hypoxia.
Effects of autophagy on endothelial cell migration in vitro
The transwell-based migration assay was established to quantitatively evaluate EAhy926 migration in vitro. As shown in Figure 4A, compared with the control group, the migration index of cells increased 123% after hypoxia treatment (p<0.05); further, in the group to which 3-MA, a widely used autophagy inhibitor, was added, the migration index increased 157% when compared with the group with only hypoxia treatment (p<0.05) (Fig. 4B). Additionally, using F-actin quantification, hypoxia could increase the formation of actin stress-fibers. Further, this effect could be dramatically increased using the autophagy inhibitor 3-MA (Fig. 4C). These results suggest that the hypoxia-induced autophagy made endothelial cells to become immobile and quiescent, and this process could be reversed by 3-MA.
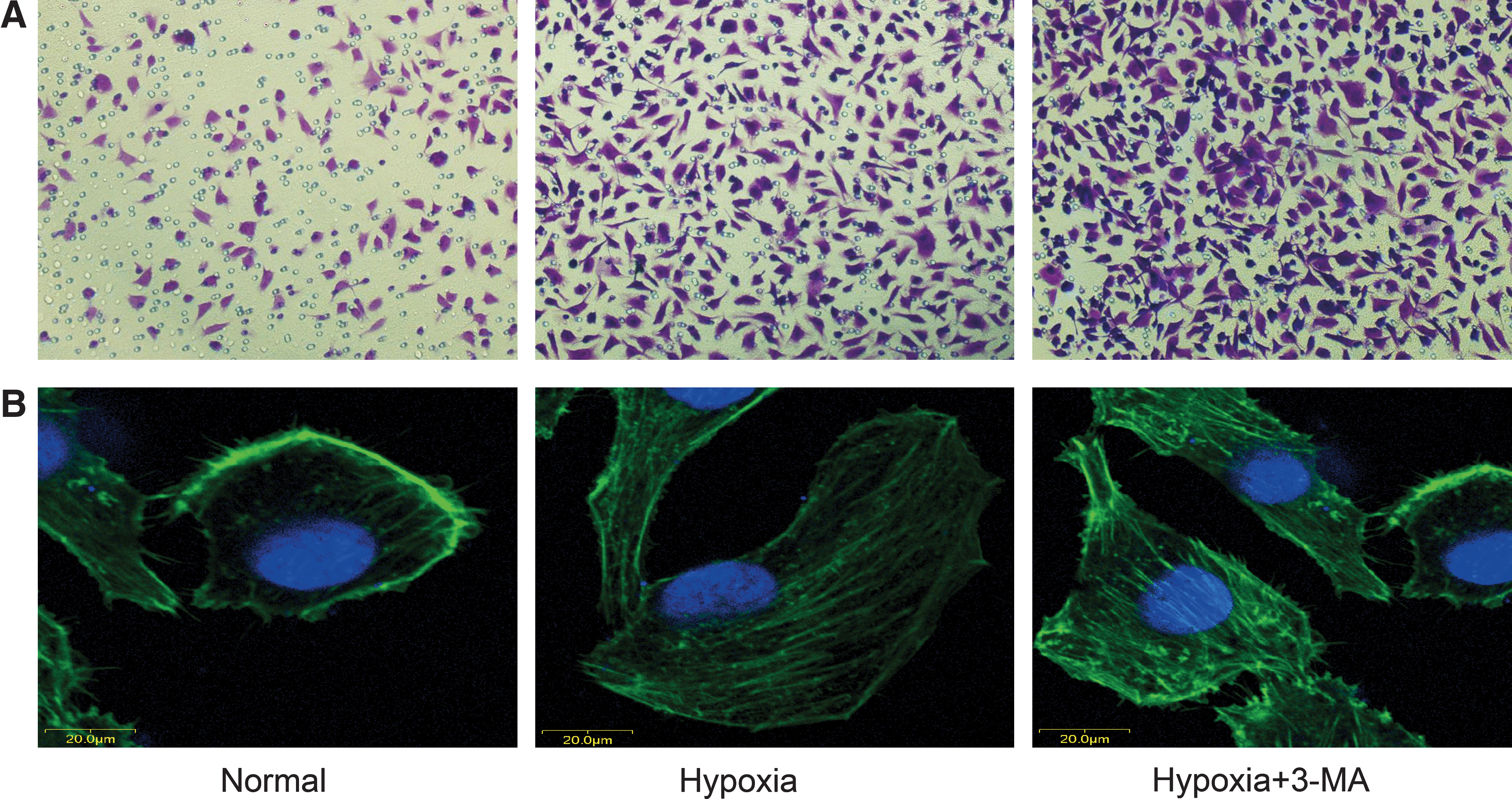
Effects of autophagy on endothelial cell migration in vitro.

Discussion
Autophagy is typically observed under extreme conditions such as amino acid deprivation and hypoxia in mammalian cells. When autophagy is induced, double-membrane structures, called autophagosomes, are derived from the endoplasmic reticulum or an unknown source sequester within the cytoplasm, often including intracellular organelles such as the mitochondrion and endoplasmic reticulum, and then merge with lysosomes, which are then called autolysosomes. Autophagosomes and autolysosomes are collectively called autophagic vacuoles. Eventually, the inner membrane disappears and the contents inside are degraded by lysosomal hydrolases. Thus, autophagy is characterized by the prominent formation of autophagic vacuoles. Previous studies have demonstrated that autophagy occurs not only under physiologic conditions and during development but also under some pathologic conditions such as degenerative diseases, infectious diseases, and cancer. 14 As the authors and others have reported, many cancer cell types undergo autophagy in response to anticancer therapies, including radiation. 15
Hypoxia-induced autophagy is a survival response. Autophagy has been characterized as a survival response as well as a pathway of cell death. This pathway is normally induced under conditions of stress such as nutritional or growth factor deprivation. 16 The present study shows that hypoxia can induce autophagy in endothelial cells independent of nutritional stress. Autophagy is an evolutionarily conserved mechanism involving the formation of autophagosomes that sequester cytoplasmic macromolecules and organelles before fusion with the endo/lysosomal compartment. 17,18 Despite recent advances in understanding its molecular mechanisms and biological function, it remains controversial as to whether autophagy acts primarily as a cell survival or cell death pathway. 19 Autophagy has been described as a form of nonapoptotic or necrotic cell death based on morphological criteria observed for a variety of cell types during development. However, in the context of hypoxia, nutrient depletion, or growth factor deprivation, it is clear that autophagy is crucial in maintaining cellular ATP production and macromolecular synthesis and, therefore, represents an essential prosurvival pathway. 20 The present study shows that hypoxia (1% O2 for 6 or 24 hours) does not kill the numerous cells tested but rather induces autophagy, a survival process. The recent work of Zhang et al. 21 revealed that hypoxia induces the autophagy of mitochondria to prevent an increase in the level of reactive oxygen species and cell death. Therefore, hypoxia appears to be an early prosurvival process, probably acting as a warning signal for cells to anticipate extreme nutritional stress.
Several signaling pathways, including the Beclin1/PI3K complex, ATG1 complex, and the p38 MAPK pathway, are involved in the induction of autophagy. 22 MAPK p38 has a dual role in autophagy, both as a positive and negative regulator. It is possible that the autophagic response may depend upon the nature of the stimulus and the strength and duration of the activated MAPK pathways. The observations in the present study are consistent with a role for Beclin1/PI3K in regulating autophagy and the autophagy-mediated hypoxia stress. Thus, the expression upregulation of Beclin1 protein was detected in endothelial cells during hypoxia stress. Inhibiting PI3K using LY-294002 decreased the levels of p-Akt, and down-regulating the expression of Beclin1 by shRNA could decrease the number of autophagosomes, thus reducing hypoxia-induced autophagy. In contrast, inhibiting p38 MAPK using SB-203580 could also decrease the expression levels of p-MAPK p38, but not the number of autophagosomes. The data indicate that Beclin1/PI3K-dependent signals can cooperate in promoting the autophagy of hypoxic stress in endothelial cells.
As sustained autophagy can lead to cell death, the pathway is also referred to as programmed cell-death type II (PCD II). 23 Alternatively, inhibiting apoptosis (PCD I) can also activate autophagy and vice versa. 24 Endothelial cells possess many redundant survival pathways under stress. For example, endothelial cells treated with chemotherapeutic agents such as carboplatin induce expression of VEGF, a survival growth factor for endothelial cells. 25 Recent studies using conditional knock-out of VEGF in endothelial cells have elucidated the importance of stress-induced autocrine stimulation of endothelial cells by VEGF. 26 The present study provides evidence for another survival mechanism, autophagy, which is initiated by the stress of hypoxia-induced endothelial cells.
Conclusions
In conclusion, the present study suggests that endothelial cells mount an autophagic survival response to hypoxia and inhibition of autophagy is required to channel them toward apoptotic cell death. Thus, concomitant inhibition of autophagy may be a useful strategy to increase the potency and efficacy of hypoxia stress and inhibit angiogenesis.
Footnotes
Acknowledgments
The authors are grateful to Marja Jäättäla (Apoptosis Department and Centre for Genotoxic Stress Research, Copenhagen, Denmark) for providing the LC3-GFP construct. The authors also thank S. Ramakrishnan (Department of Pharmacology, University of Minnesota Medical School, Minneapolis, MN) for providing Beclin1 shRNA. This work was supported by the National Natural Science Foundation of the People's Republic of China (No. 30973473/H1611).
Disclosure Statement
There are no financial conflicts.