
Abstract
CK2 interacts and phosphorylates >300 proteins, including Stat3, and is linked to a number of human cancers. Constitutively activated Stat3 has been reported in 50% of human lung cancers. Inhibition of CK2 activity can induce apoptosis and suppression of Stat3 activation in cancer cells. This study examined the effects of CK2 inhibitors on growth inhibition of lung cancer cells and the therapeutic potential on lung cancer. The CK2 inhibitor and radiation both suppressed cancer cell growth in a dose-dependent manner. Besides, the cytotoxic effect of irradiation could be augmented by CK2 inhibitors (p<0.05, two-way analysis of variance and Tukey's Honestly Significant Difference). Moreover, the growth inhibition of CK2 inhibitor and irradiation was both associated with suppression of Stat3 activation. Taken together, inhibition of CK2 activity appears to be a promising treatment strategy for non-small cell lung cancer and CK2 inhibition results in reduced Stat3 activation. Our data warrant further effort to develop CK2-targeted radiosensitizer for lung cancer treatment.
Introduction
CK2 is a pleiotropic serine/threonine protein kinase phosphorylating and interacting with multiple cellular proteins. CK2 is a heterotetramer composed of 2 catalytic, namely, CK2α (42–44 kDa) and/or CK2α′ (38 kDa) (α and/or α′), and two regulatory β-subunits. 1,2 CK2 phosphorylates and interacts >300 proteins that involve in major cellular processes, including replication, transcription, translation, signal transduction, and cell death. 2 –4 Overexpression of CK2 has been documented in a number of cancers, including the kidney, mammary gland, lung, head and neck, and prostate malignancies. 5 –9
Downregulation of CK2 activity by means of chemical inhibition, as well as antisense oligodeoxynucleotide, RNAi, or overexpression of exogenous kinase-inactive CK2 can induce apoptosis in cancer cells. 10 –14 Chemical inhibitors can penetrate the cells easily and presumably are able to inhibit all CK2 in cells. CK2 inhibitors of different classes of chemical compounds have been investigated as ATP competitive inhibitors of CK2. 15 4,5,6,7-Tetrabromo-1H-benzotriazole (TBB) and its derivatives are among one of the four major classes of CK2 inhibitors. TBB was ever demonstrated to inhibit other protein kinases although with less potency than CK2, including CDK2, GSK3β, and DYRK1a. 16 Tetrabromocinnamic acid (TBCA), a derivative of TBB with better CK2 selectivity, has been confirmed to have no cross activity on DYRK1a. 17
Stat3 is constitutively activated in about 50% of non-small cell lung cancer (NSCLC) primary tumors and lung cancer cell lines. 18 –20 Inhibiting Stat3 increases apoptosis of NSCLC cells. 18,21 Autocrine IL-6-induced Stat3 activation has been reported to be associated with the pathogenesis of lung adenocarcinoma and activating epidermal growth factor receptor (EGFR) mutation in lung adenocarcinoma. 22,23 Evidence has shown that CK2 inhibition resulted in impaired IL-6-dependent Stat3 phosphorylation at Tyr702 and Ser727 in multiple myeloma cells. 24 Therefore, CK2 inhibitors may suppress lung cancer growth through inhibition of Stat3 signaling.
Radiotherapy and concurrent chemoradiotherapy are commonly used in lung cancer treatment for either curative or palliative purposes. Ionizing radiation induces cell cycle arrest at the G1/S and G2/M checkpoints. Concurrent chemotherapy can act as a radiosensitizer. 25 Radiosensitization induced by Cisplatin acts through enhanced formation of toxic platinum intermediates in the presence of radiation-induced free radicals, inhibition of DNA repair, radiation-induced cellular platinum uptake, and cell cycle arrest. 26 Gemcitabine inhibits ribonucleotide reductase to deplete deoxyadenosine triphosphate pool and synchronize cells in S phase. 26 Paclitaxel and Vinorelbine inhibit mitosis, resulting in accumulation of cancer cells in G2/M. 27,28
Stat3 is a well-known key element controlling the G1 to S cell cycle transition. 29 Kim et al. observed that phosphor-Stat3 Tyr705 levels were decreased in human umbilical vein endothelial cells after irradiation, but not in breast cancer cells. 30 Besides, uncontrolled Stat3 signaling antagonized G1 arrest and conferred inherent radioresistance. 31,32 Moreover, knocking down of Stat3 by an antisense oligonucleotide enhanced radiation-induced apoptosis in prostate cancer cells. 33 In addition, CK2 is required for the cell cycle transition through G0/G1, G1/S, and G2/M as well. 34 Therefore, suppression of CK2 activity may enhance radiosensitivity of cancer cells through Stat3 inhibition.
This study demonstrates that CK2 inhibitors suppressed growth and enhanced the radiosensitivity of lung cancer cells and the correlation of CK2 inhibition with reduced Stat3 phosphorylation. Our data reveal a potential therapeutic application of CK2 inhibitors in cancer treatment and shed a light on overcoming radioresistance of human cancers.
Materials and Methods
Human non-small lung cancer cell lines
Human lung cancer cell lines with wild-type EGFR, A549 (CCL-185, bronchoalveolar carcinoma) and NCI-H460 (HTB-177, large cell carcinoma), and EGFR mutants, NCI-H1650 (CRL-5883, adenocarcinoma) and NCI-H1975 (CRL-5908, adenocarcinoma) were purchased from American Type Culture Collection (ATCC, Manassas, VA). H460 and A549 were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. H1650 and H1975 cells were cultured in RPMI-1640 complete medium containing 10% fetal bovine serum. Culture media were supplemented with 10 units/mL penicillin and 10 μg/mL streptomycin. All cells were cultured at 37°C in a humid incubator with 5% CO2.
CK2 inhibitors and reagents
TBB and TBCA ((E)-3-(2,3,4,5- Tetrabromophenyl)acrylic acid) were purchased from Calbiochem® Biochemicals (Darmstade, Germany). Hematein was purchased from Timtec Inc. (Newalk, DE) and prepared as described previously. 35 IC50 of CK2 inhibitors for cancer cells were titrated with different concentrations (described in Cell Growth and Viability section). Recombinant human EGF was obtained from Invitrogen (PHG0311; Carlsbad, CA).
Western blotting
Treated cells were washed twice with PBS, scraped off the plates, and lysed in cell lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate [SDS]). Whole cell lysates were boiled and the protein concentrations were determined with the Bradford assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein (20–40 μg) were separated by SDS-polyacrylamide gel electrophoresis under reducing conditions in 12%–15% linear gradient polyacrylamide gels. After transferring the proteins onto Immobilon-P (Millipore, Billerica, MA), the membranes were blocked with 5% nonfat milk powder and 0.2% Tween 20 in Tris-buffered saline overnight at 4°C and then incubated with the primary antibody for 1 hour at room temperature. Membranes were washed in Tris-buffered saline for three 5-minute periods. Primary antibody for CK2α was obtained from Abnova (Taipei, Taiwan). Stat3, phosph-Stat3 (pStat3, Tyr705), Akt, and Phospho-Akt (pAkt, Ser473) antibodies were from Cell Signaling Technology (Danvers, MA). γ-tubulin antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies were used as secondary antibodies (Santa Cruz Biotechnology Inc.). Proteins were observed with chemiluminescence luminal reagents (Santa Cruz Biotechnology Inc.). Densitometric strength of blots was calculated by using ImageJ (v1.44m for Windows, National Institutes of Health).
Cell growth and viability
About 3000 cells/well were seeded onto 96-well plate and incubated overnight. Cells then were treated with CK2 inhibitors for 48 hours. Growth inhibition of treated cells was determined by using the colorimetric MTT cell viability/proliferation assay (Sigma-Aldrich, St. Louis, MO) according to manufacturer's recommendations. Briefly, cells were treated with MTT reagent and allowed to react for 1.5 hours at 37°C before adding the solubilization reagent (100 μL 20% SDS in 50% dimethyl formamide and 50% H2O [pH 4.7]; pH adjusting solution, 80% acetate and 20% 1M HCl). Substrate cleavage was monitored at 570 nm. The viability of cells was assessed by morphology analysis using an inverted phase-contract microscope and trypan blue exclusion assays.
CK2 inhibitors on EGF-induced Stat3 activation
Cells (5×105/well) were plated in 6-cm plates overnight. After replacement with serum-free culture media, cells were incubated with TBB 10 μM or TBCA 25 μM or vehicle (dimethyl sulfoxide) for 2 hours, and were subsequently treated with 100 ng/mL EGF for 30 minutes. Cells were collected at 0, 5, 20, and 60 minutes for Western blotting of total and phosphorylated Stat3 (Tyr705).
Irradiation
Cells (1×105/well) were plated onto six-well culture plates and incubated overnight at 37°C. Next, cells were treated with CK2 inhibitors (0, 12.5, 25, and 50 μM for TBB and TBCA; 0, 50, 100, 200, and 400 μM for hematein). Dimethyl sulfoxide (0 μM) served as the negative control. Forty-eight hours after treatment, cells were irradiated (0, 2, 4, or 8 Gy in a single fraction) with a 6-MeV electron beam generated by a linear accelerator (Clinac 21EX; Varian, Palo Alto, CA) at a dose rate of 300 cGy/min. On day 3 after irradiation, triplicate cultures for each combination treatment were counted for viable cells. For Western blotting, cells were treated with 50 μM TBB for 2.5 hours before irradiation with dose of 4 Gy. About 30 minutes after irradiation, cells were collected for protein extraction and further analysis.
Statistical analysis
Results are given as means±standard deviations (SD). Student's unpaired t-test was used to compare different treatments. Two-way analysis of variance (ANOVA) was used to compare the survival proportions and SD at the different CK2 inhibitors concentrations and varying radiation doses for treating lung cancer cells. Pair-wise comparisons were made using Tukey's Honestly Significant Difference test. A p-value <0.05 was considered as statistically significant. Data were analyzed with Stata statistical software version 10.1 (StataCorp, College Station, TX).
Results
CK2α is ubiquitously expressed in lung cancer cells
To understand the role of CK2α in lung cancer, the CK2α expression in lung cancer cells was first examined by Western blotting. Of note, H460 and A549 cells harbor wild-type EGFR, whereas H1650 and H1975 cells are EGFR mutants. As shown in Figure 1, CK2α was expressed in all tested cells unrelated to the status of EGFR mutation. H1650 cells had the highest CK2α expression and the H838 cells got the lowest level.

CK2α expression in human lung cancer cells. The CK2α is ubiquitously expressed in lung cancer cell lines (upper band). Mouse monoclonal antibody (clone 3D9) was obtained from Abnova Corporation. Number below each lane denoted the CK2α: γ-tubulin ratio.
CK2 inhibitors suppress growth of lung cancer cells
Then tested was the effect of CK2 inhibitors on lung cancer cells with different concentrations of TBB, TBCA, and hematein for dose titration. All CK2 inhibitors suppressed growth of lung cancer cells in a dose-dependent manner (Fig. 2). Notably, among three CK2 inhibitors, the dose needed to suppress cancer cell growth was mostly higher for hematein (Fig. 2). The IC50 of each CK2 inhibitors for cells was summarized in Table 1. Remarkably, A549 cells were more susceptible to CK2 inhibitors compared with other cell lines examined.

Growth inhibition of lung cancer cells by CK2 inhibitors. Cells (3000/well) were seeded into 96-well plates. After incubation overnight, cells were treated with CK2 inhibitors with various concentrations for 48 hours. All CK2 inhibitors suppressed lung cancer cell growth. *, **, *** denote the lowermost concentrations with significant growth inhibition (p<0.05) compared with 0 μM (dimethyl sulfoxide) for TBB, TBCA, and hematein, respectively. TBCA, tetrabromocinnamic acid; TBB, 4,5,6,7-tetrabromo-1H-benzotriazole.
TBB, 4,5,6,7-tetrabromo-1H-benzotriazole; TBCA, tetrabromocinnamic acid.
CK2 inhibitors block EGF-induced Stat3 activation
The effect of CK2 inhibitors on Stat3 activation was examined. Cells were pretreated with 25 μM TBB or TBCA for 2 hours before EGF stimulation. Comparing with A549 cells, H1975 cells had a higher portion of activated Stat3 in baseline (mock). Pre-treatment with TBB or TBCA did not inhibit the pStat3 in unstimulated A549 cells. No difference was noted in pStat3 before EGF stimulation in A549 cells. Upon EGF stimulation, Stat3 activation was suppressed by 25 μM TBB or TBCA, whereas the total Stat3 remained unchanged in A549 cells. Contrarily, CK2 inhibitors suppressed baseline Stat3 activation in H1975 cells. There was no significant Stat3 activation after EGF stimulation in mock-treated H1975 cells. However, pStat3 increased after EGF stimulation in H1975 cells pretreated with CK2 inhibitors (Fig. 3).
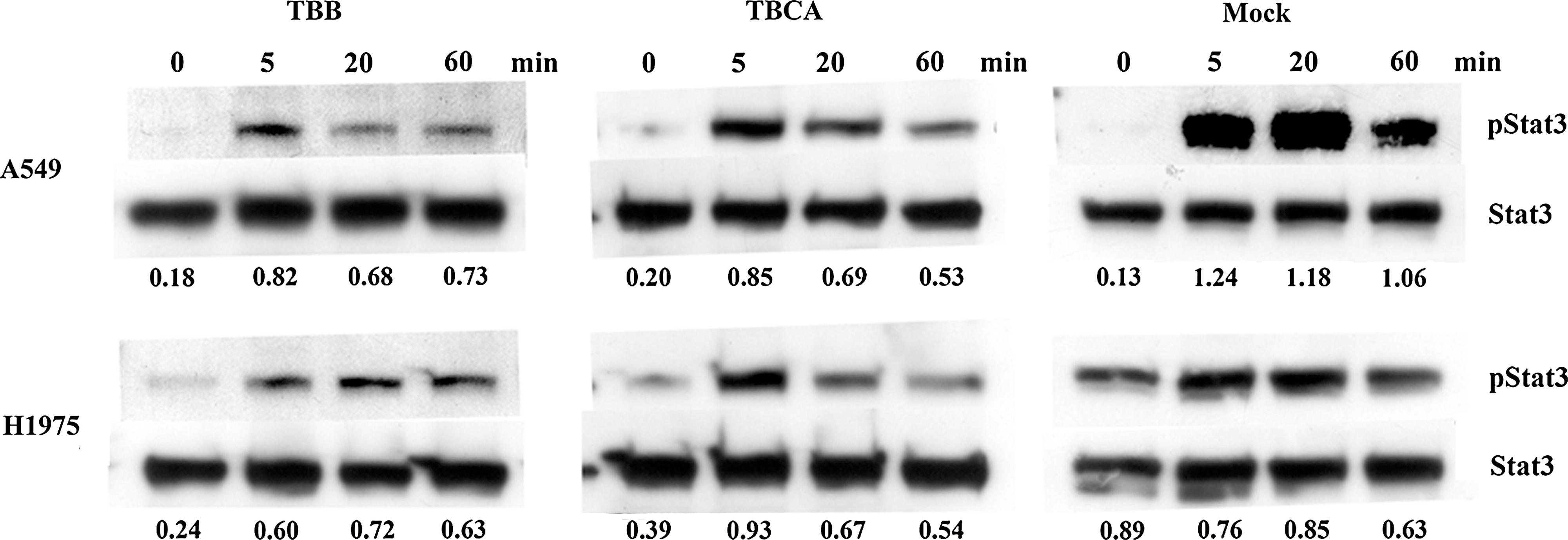
CK2 inhibitors suppressed EGF-induced Stat3 activation in lung cancer cells. Cells (500,000) were seeded in 6 cm2 plate overnight. Cell were treated for 2 hours with 25 μM TBB or 25 μM TBCA and stimulated with 100 ng/mL EGF for 0, 5, 20, and 60 minutes. Number below each lane denoted the pStat3:Stat3 ratio.
CK2 inhibitors enhance the radiosensitivity of NSCLC cells
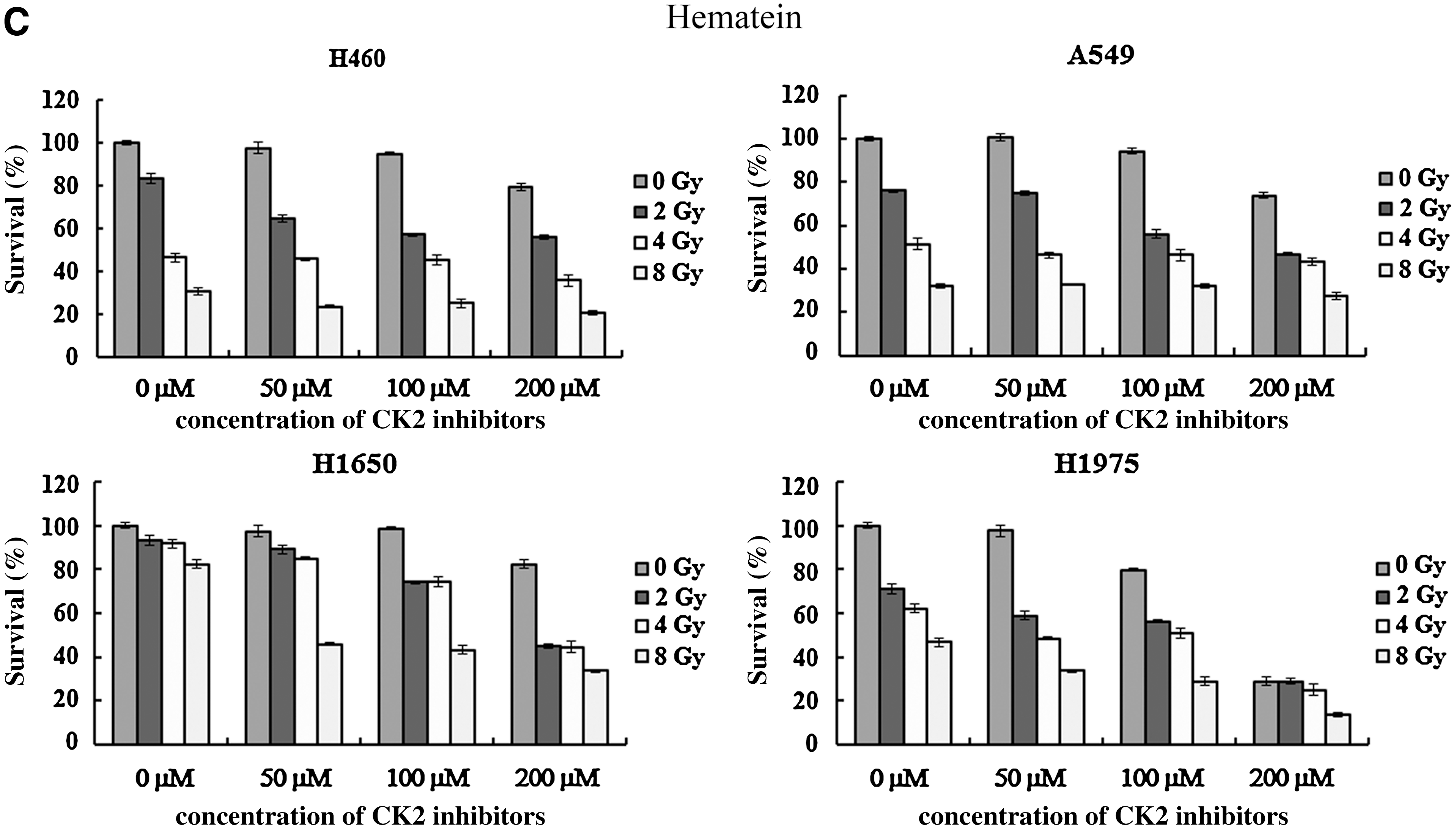
Various doses of CK2 inhibitors combined with different radiation doses (0–8 Gy) were applied for cancer cell treatment. Whether or not combined with irradiation, a higher concentration of CK2 inhibitors resulted in a lower survival proportion of lung cancer cells (two-way ANOVA, p<0.05). Survival of cancer cells was dose-dependent to radiation doses, irrespective of the form and concentration of CK2 inhibitors (two-way ANOVA, p<0.05; Fig. 4A–C). Combination of CK2 inhibitor and radiation was more toxic than CK2 inhibitor or radiation alone (two-way ANOVA, p<0.05). Results were further confirmed by Tukey Honestly Significant Difference pair-wise comparisons. Comparing to TBB and TBCA, a higher dose of hematein was required to inhibit growth of lung cancer cells. High dose of CK2 inhibitors, 50 μM TBB/TBCA or 400 μM hematein, was too toxic to cancer cells when combined with radiation (data not shown).
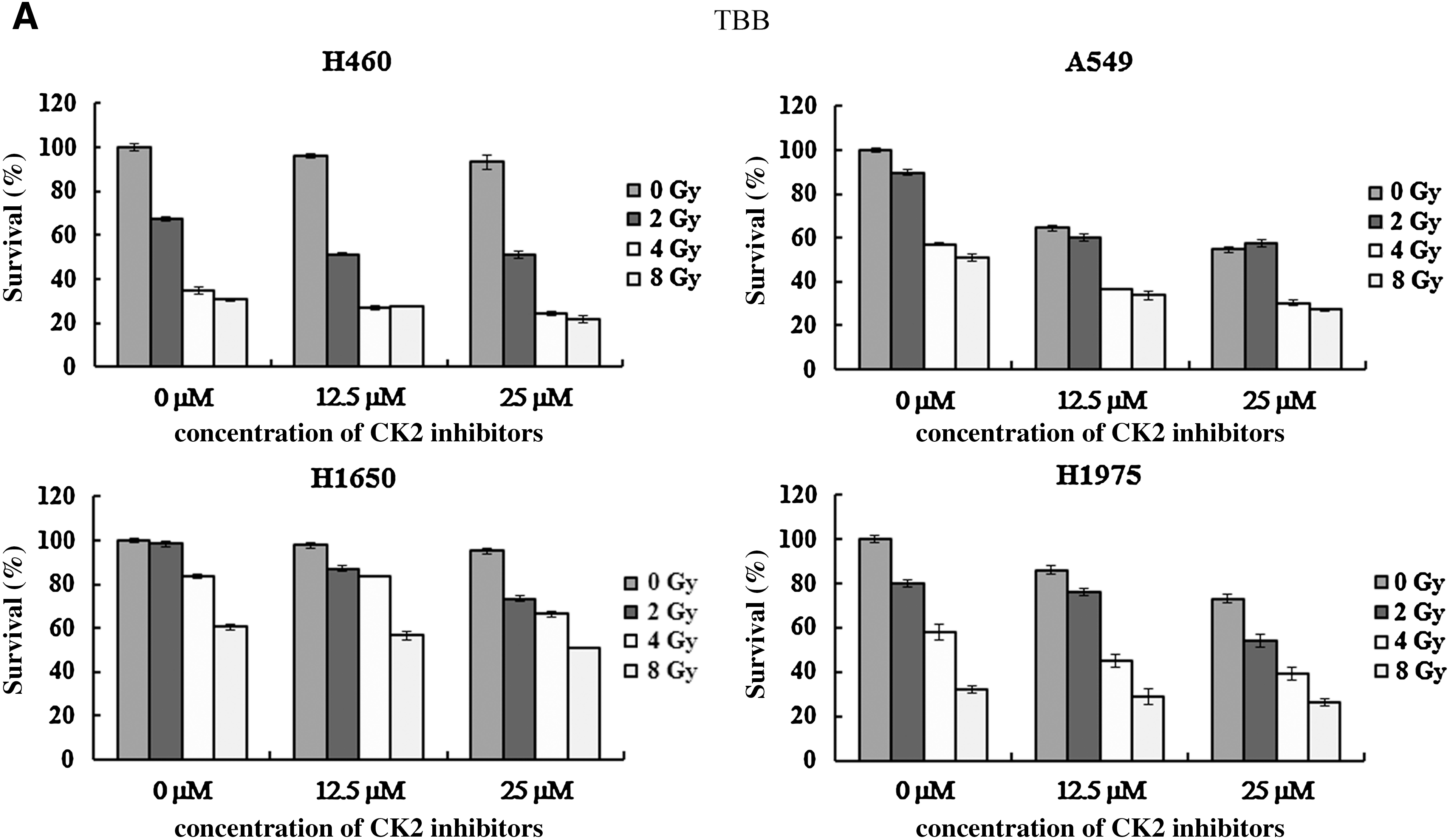
Combination of CK2 inhibitors and irradiation in lung cancer cells.

CK2 inhibitors and radiation suppress Stat3 activation
Since Stat3 is related to cell growth and proliferation and CK2 inhibitor can suppress Stat activation, it is intriguing to know if the tumor suppression induced by CK2 inhibitor and/or irradiation is associated with Stat3 activity. Therefore, pStat3 was examined under CK2 inhibitor, irradiation, and combination of both. As shown in Figure 5, activated Stat3 was reduced by CK2 inhibitor and irradiation and, to a greater extent, by combination of CK2 inhibitor and irradiation, but in terms of Stat3 activation as expressed by pStat3:Stat3 ratio, the combination of CK2 inhibitor and irradiation resulted in synergistic effect in H460 and A549 cells but antagonistic effect in H1650 and H1975 cells (Table 2). Meanwhile, the total Akt and activated Akt remained unchanged under treatment with CK2 inhibitor and/or irradiation.

CK2 inhibitor and irradiation inactivated Stat3. Cells (1×105/well) were plated onto six-well culture plates and incubated overnight at 37°C. Cells were then treated with 50 μM TBB for 2.5 hours before irradiation with dose of 4 Gy. Thirty minutes after irradiation, cells were collected for protein extraction and further analysis.
DMSO, dimethyl sulfoxide.
Discussion
CK2 is ubiquitously expressed in cancer cells, indicating the importance of CK2 in cancer cell homeostasis (Fig. 1). CK2 phosphorylates and interacts proteins involving in cellular processes including replication, transcription, translation, signal transduction, and cell death. 2 –4 Overexpression of CK2 has been documented in a number of human cancers, including lung cancer. 5 –9 Hence, it is a reasonable approach to suppress cancer cell through CK2 inhibition.
Lung cancer cells were first treated with CK2 inhibitors and found that CK2 inhibitors suppressed growth of cancer cells dose dependently (Fig. 2). Among tested cells, A549 cells were more vulnerable to CK2 inhibitors. Phosphorylated Stat3 in two cancer cell lines, A549 with wild-type EGFR and H1975 with an activating EGFR mutation, was examined. CK2 inhibitors blocked EGF-induced Stat3 activation, whereas the total Stat3 remained unaffected in A549 cells. Contrarily, the CK2 inhibitors had no effect on pStat3 in H1975 cells (Fig. 3). Besides, baseline pStat3 was higher in H1975 cells. The higher baseline pStat3 in H1975 cells is possibly due to the activating EGFR mutation, which leads to constitutively activated Stat3 and resistant to treatment with CK2 inhibitors. 36,37
The dose of CK2 inhibitors used in our test may be insufficient. The concentration of 25 μM is about half of the TBB/TBCA IC50 for H1975 cells (Table 1). Hence, 25 μM TBB/TBCA may not be able to suppress EGF-stimulated Stat3 phosphorylation in H1975 cells. Nevertheless, 25 μM TBB or TBCA sufficiently suppressed baseline pStat3 in H1975 cells. Whether the different effects in A549 and H1975 cells are correlated with intrinsic EGF signaling demands further studies.
In this study, the growth of lung cancer cells was suppressed by CK2 inhibitors and/or irradiation (Fig. 4A–C). Remarkably, the effect of cancer cell killing by radiation was further augmented by adding CK2 inhibitors (p<0.05). Therefore, CK2 inhibitors can enhance the radiosensitivity of lung cancer cells. Further, the effect of cancer killing was correlated with suppression of pStat3 (Fig. 5).
Stat3 is a well-known oncogene that regulates cell cycle and averts apoptosis. 38 In addition, activated Stat3 signaling confers inherent radioresistance in malignancies. 32 Stat3 phosphorylation was reduced in cancer cells treated with TBB or irradiation. Synergistic suppression of pStat3 could be achieved by combination of radiation and TBB in H460 and A549 cells (Fig. 5). In contrast, antagonistic effect on suppression of Stat3 activation by combining radiation with TBB was found in H1650 and H1975 cells. Whether the different effects are due to presence of activating EGFR and/or other unknown factors remained unanswered in this study. Nevertheless, the above findings indicate that CK2 inhibitors enhance the radiosensitivity of lung cancer cells through inhibition of Stat3 phosphorylation.
Since CK2 interacts with >300 proteins, 2 –4 CK2 inhibitors may act through other pathways as well as Stat3 suppression to inhibit growth and augment radiosensitivity in lung cancer cells. The phosphatidylinositol-3-kinase (PI3-K)/Akt pathway is known to be associated with radiation resistance mechanisms. 39 In NSCLC, phosph-Akt (pAkt) at Ser473 was correlated with radioresistance; inhibition of Ser473 pAkt increased radiation-induced apoptosis of lung cancer cells. 40 Besides, CK2 inhibition could suppress Ser473 pAkt in acute myeloid leukemia cell. 41 In this study, however, pAkt (Ser473) could not be suppressed with CK2 inhibitors, irradiation, or both. Whether irradiation-induced Akt activation and/or simply the inappropriate timing for protein collection count for failure to suppress Ser473 pAkt necessitates further study.
Taken together, our data demonstrate that CK2 inhibitors can suppress the growth and enhance the radiosensitivity of lung cancer cells by suppression of Stat3 activity. Inhibition of CK2 activity appears to be a promising strategy for NSCLC treatment and sheds a light on overcoming radioresistance of human cancers. Our data deserve further efforts to develop CK2-targeted radiosensitizer for lung cancer treatment.
Footnotes
Acknowledgments
This study was supported by grants CMRPG670411 to C.-T. Yang and CMRPG670241 to Y.-C. Lin from Chang Gung Memorial Hospital. The authors thank Dr. H.-H. Weng at Chang Gung Memorial Hospital, Chiayi branch, for his generous aid in statistical analysis.
Disclosure Statement
No competing financial interests exist.