
Abstract
The importance of apoptosis during the process of inhibiting tumorigenesis has been recognized. The role of BH3-only proapoptotic protein Bcl-2–associated death (BAD) in tumor growth remains controversial. The aim of this study was to explore the role of BAD in lung cancer cells. Our study showed that expression of BAD was upregulated in A549 cells by a recombinant lentivirus overexpressing BAD. In vitro, BAD overexpression significantly inhibited A549 cell proliferation and induced apoptosis in cell proliferation and apoptosis assays, respectively. The effect of BAD on A549 cells was studied in tumor xenograft of nude mice and the results showed that the tumor volume in the experimental group was smaller than the control groups. Further, immunohistochemical technique was used to determine the cell proliferation and apoptosis status of the lung tumor xenograft cells. This demonstrated that the in vivo and in vitro results were consistent. Taken together, our results indicate that overexpression of BAD inhibits the growth of A549 cells in vitro and in vivo, through inhibiting cell proliferation and inducing apoptosis. Thus, BAD could be a potential therapeutic target.
Introduction
Lung cancer is one of the most common malignant tumors in the world with the highest incidence and mortality. Lung and bronchial cancer alone accounted for 15% (116,750) and 14% (105,770) of all newly diagnosed cancers in men and women, respectively. In addition, lung and bronchial cancer was the leading cause of cancer deaths among men and women in the United States in 2010. 1 Latest data from China showed that lung cancer accounted for 27.29% (4,098,000) of cancer deaths in urban areas and 19.90% (2,571,000) in rural areas in 2004–05. Additionally, lung cancer was found to be the leading cause of cancer deaths in the current decade in China. 2 Further, the early symptoms of lung cancer are nonspecific; therefore, it is often diagnosed at an advanced stage, this together with its refractory nature to conventional chemotherapy gives it a poor prognosis. 3 The treatment of lung cancer has reached a plateau. To reduce morbidity and mortality from the disease, new therapeutic strategies for advanced cancer in addition to primary prevention and early diagnosis are essential.
Apoptotic pathways frequently exert a critical role in both tumor progression and drug resistance. Therefore, proteins associated with this pathway have the potential to be therapeutic targets. 4 Bcl-2–associated death (BAD) protein is one of the Bcl-2 family members, which are key regulators of apoptosis. The Bcl-2 family has >20 members in mammals. They control cell survival or death by regulating mitochondrial function. 5 More and more studies have demonstrated that the antiapoptotic Bcl-2 family members are prone to be oncogenes. Conversely, proapoptotic members including BAD are likely to be tumor suppressors.
Extensive investigations have uncovered BAD to be a promising protein for helping to identify potential targets for lung cancer therapy. 6 First, Berrieman et al. have demonstrated that BAD is expressed in normal lung tissue, but loss of BAD expression was observed in 51% of primary nonsmall cell lung carcinoma (NSCLC) samples. 7 It suggested a role for loss of expression of BAD in facilitating the development of NSCLC. Second, overexpression of BAD blocked BCL-XL–dependent cell survival 8 and resulted in apoptosis of mammalian cells. 9,10 Finally, previous studies have indicated that BAD activation specifically lowered the mitochondrial threshold for cell death 11 and led to mitochondrial dysfunction and cell apoptosis in lung adenocarcinoma cells. 12 Thus, the current study was designed to examine the effects of BAD on human A549 lung adenocarcinoma cells.
Materials and Methods
Tumor cell line and cell culture
Human A549 lung adenocarcinoma cells and 293T cells conserved in the Laboratory of Stem Cell Biology were maintained in DMEM (Thermo), containing 10% fetal bovine serum (Thermo) and 1% penicillin/streptomycin (Thermo), in a humidified 5% CO2 incubator at 37°C.
Plasmid construction and lentivirus package
The lentiviral vector (pLVX-IRES-ZsGreen1) (Clontech) was used to construct the PLV-BAD plasmid. The human BAD sequence (NM_032989) 10 was amplified by polymerase chain reaction (PCR) using the following primer: 5′-CGGAATTCATGTTCCAGATCCCAGAGTTTGA-3′ and 5′-CGGGATCCTCACTGGGAGGGGGCG-3′. The PCR products were then linked to the pGEM-T Easy Vector for sequence analysis and digested by EcoR I and BamH I before being inserted into the lentiviral vector. The package of lentivirus is as previously described. 13 The PLV-BAD plasmid or empty vector plasmid and its packaging plasmid mix (Clontech) were cotransfected using Lipofectamine 2000 (Invitrogen). After 48 hours, supernatants were collected from these cells and passed through a 0.45 μm filter (Millipore). The titer of virus was measured according to the manufacturer's instruction. A549 cells were infected with lentivirus at an MOI of 80; the infection efficiency measured by inverted fluorescence microscope was about 100%.
Western blotting
Cells were rinsed twice with phosphate-buffered saline (PBS, pH 7.2) and the lung tumor xenograft tissues were grinded in liquid nitrogen. They were then lysed in RIPA buffer as described previously, 13 and finally centrifuged at 13,000 r.p.m. for 15 minutes at 4°C. The protein was separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. After blocked with 5% nonfat milk in tris-buffered solution containing 0.1% Tween 20 for 1 hour, the membranes were incubated overnight at 4°C with anti-BAD-specific antibodies (Cell Signaling Technology) diluted in blocking buffer. This was followed by incubation with the relevant secondary antibodies (Cell Signaling Technology). The immunoreactive bands were visualized using enhanced chemiluminescence (Cell Signaling Technology). For equal protein loading, a monoclonal anti-β-actin antibody (Millipore) was used as a control.
Cell proliferation assays
Cell proliferation was measured using a Cell Counting kit (CCK)-8 (Dojindo) in 96-well plates according to the manufacturer's instructions. 14 CCK-8 was added to the wells (10 μL per well) at the experimental period. After 1.5 hours of incubation at 37°C in 5% CO2, the absorbance of each well at 450 nm wavelengths was noted using an enzyme-linked immunosorbent assay reader (Bio-Rad). Triplicate wells were used for each data point.
Cell apoptosis assays
Annexin V–allophycocyanin (APC)/7-amino-actinomycin D (7-AAD) staining was performed according to the manufacturer's instructions (BD Biosciences). 15 Cells were washed by PBS twice, resuspended in binding buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl; and 2.5 mM CaCl2), and stained with 5 μL of annexin V–APC and 5 μL 7-AAD. After 15 minutes, the cells were analyzed by flow cytometry using an FACSDiva software (BD Biosciences). Viable, unstained cells in untreated samples were used as negative controls. Annexin V has been demonstrated to bind to phosphatidylserine, which is exposed on the outer surface of cell membranes, to detect early apoptotic cells. On the other hand, 7-AAD is known to intercalate in fragmented DNA to detect late apoptotic cells. Dead cells were decided as annexin V–APC+ and 7-AAD+, apoptotic cells were defined as annexin V–APC+ and 7-AAD−, and viable cells were determined as annexin V–APC− and 7-AAD−. 16
Subcutaneous implantations
BALB/c-nu mice (SPF grade) aged 4–6 weeks were obtained from the Experimental Animal Center of Sichuan University (Chengdu). All nude mice were implanted subcutaneously with 0.1 mL of 5×106 A549 cells in solution on the right flank. 17 The mice were divided into three groups: (a) implanted A549 cells overexpressing BAD (BAD group), (b) implanted A549 cells infected with lentivirus carrying empty vector (EV group), and (c) implanted untreated A549 cells (A549 group). The latter two groups acted as controls. All the animal experiments were approved by the Animal Center's Animal Care and Use Committee of Sichuan University.
Immunohistochemistry
Immunostaining was performed as previously described. 18 Antibody staining was performed on histological sections of formalin-fixed lung tumor xenografts. The primary antibody and the dilution used for immunohistochemistry were Ki67 and 1:75 (Dako). Ki67 is considered to be an objective indicator which could reliable and comprehensive response to cell population proliferation. 19 In each tumor tissue section, five high-power fields (400×) were randomly chosen and analyzed. The labeling index for the proteins of Ki67 in each case was determined according to the formula: (number of positive tumor cells/total number of tumor cells expressed)×100%. 20
Apoptosis analysis by TUNEL staining
Apoptosis analysis of lung tumor xenografts was performed using the Terminal Deoxynucleotide Transferase dUTP Nick End Labeling (TUNEL) assay to detect DNA fragmentation according to the manufacturer's instructions (Roche). Every tumor tissue section was analyzed in five randomly chosen high-power fields (400×). The apoptotic index shows the ratio of apoptotic cells to the total number of cancer cells. 17
Statistical analysis
Statistical analysis was done using SPSS 13.0 for Windows. Data were shown as mean±standard deviation. Statistical analysis of the data was performed using the one-way analysis of variance test. A p-value of < 0.05 was considered as statistically significant.
Results
BAD overexpression inhibits tumor cell growth and induces tumor cell apoptosis in vitro
As shown in Figure 1A, Western blotting demonstrated that infection with a recombinant lentivirus overexpressing BAD led to a significant increase of BAD expression in A549 cells with the same level of β-actin.

Schematic representation of the BAD expression in the three groups and BAD overexpression inhibits tumor cell growth and induces tumor cell apoptosis in vitro.
In subsequent studies, a noticeably slower growth rate was observed in the BAD group compared with the control groups, when the cell growth in a synchronous culture was assayed using CCK-8 uptake. 21 At 144 hours, cell growth was suppressed by 40%–50% more in BAD group than the A549 and EV groups (p<0.05). It was also noticed that in the BAD group, there was a phase of slow growth between 96 and 144 hours, compared with the A549 and EV groups, and the difference is statistically significant (p<0.05) (Fig. 1B).
Additionally, overexpression of BAD resulted in apoptosis of A549 cells in vitro. Apoptosis was determined by annexin V–APC and 7-AAD staining. As shown in Figure 1C, 3.7% and 3.3% of cells were demonstrated to be apoptotic cells in the A549 and EV groups, respectively. In the BAD group, 21.5% of cells were shown to be apoptotic cells. This is an increase of greater than fivefold and sixfold when compared with the EV and A549 groups, respectively.
BAD overexpression inhibits growth and induces apoptosis of tumor cells in vivo
As shown in Figure 2A, Western blotting demonstrated that tissues of lung tumor xenograft, which was implanted with A549 cells overexpressing BAD, had a higher expression of BAD with the same level of β-actin compared with the EV and A549 groups.
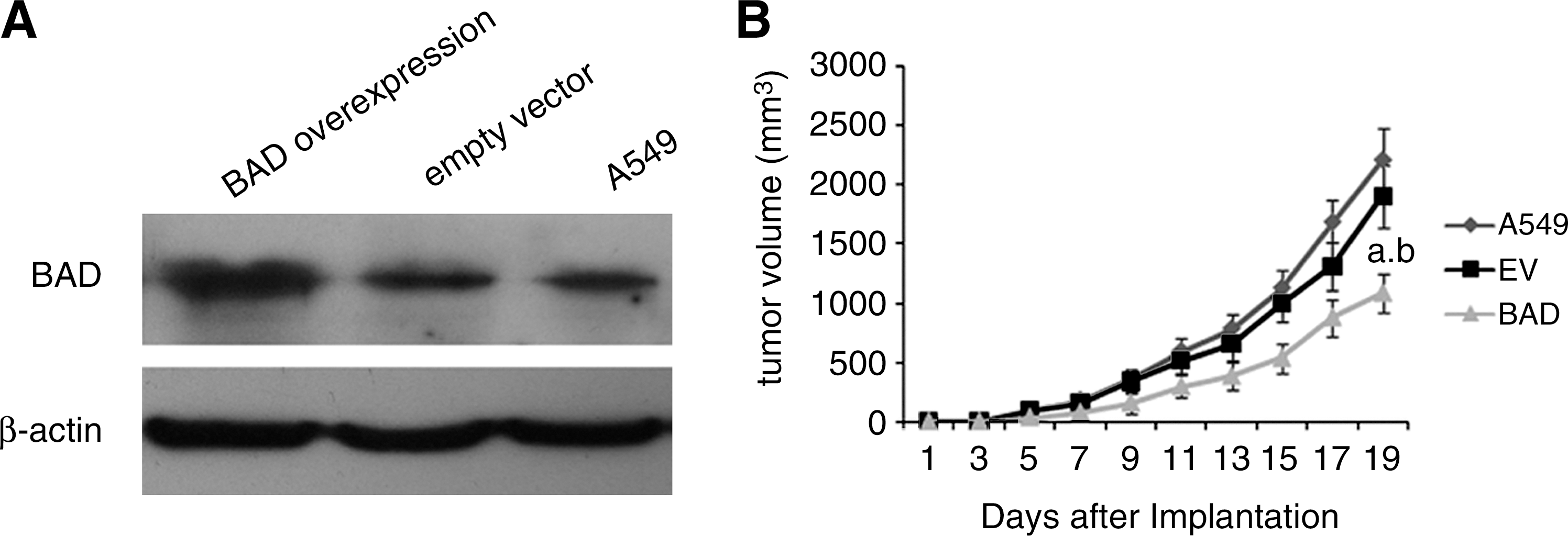
Schematic representation of BAD protein level of lung tumor xenograft tissues and BAD inhibiting tumor cell growth in vivo.
To determine whether overexpression of BAD inhibits lung tumor growth in vivo, the growth of the lung tumor xenografts in the three groups was compared. Measurement of the tumor volumes began once the subcutaneous tumors became palpable. This continued until the tumor was excised on day 19. The results showed that the xenografts in the BAD group had lower tumor volumes (1081.06±161.55) on day 19 than both the xenografts in the EV (1897.64±266.67) (p<0.01) and A549 groups (2207.36±259.79) (p<0.01) (Fig. 2B).
To investigate how BAD overexpression may have reduced the tumor volume in vivo, tissue sections of lung tumor xenograft were stained with Ki67 to assess the rate of tumor cell proliferation. The BAD group showed reduced rates of tumor cell proliferation when compared with the EV and A549 groups (p<0.05) (Fig. 3).
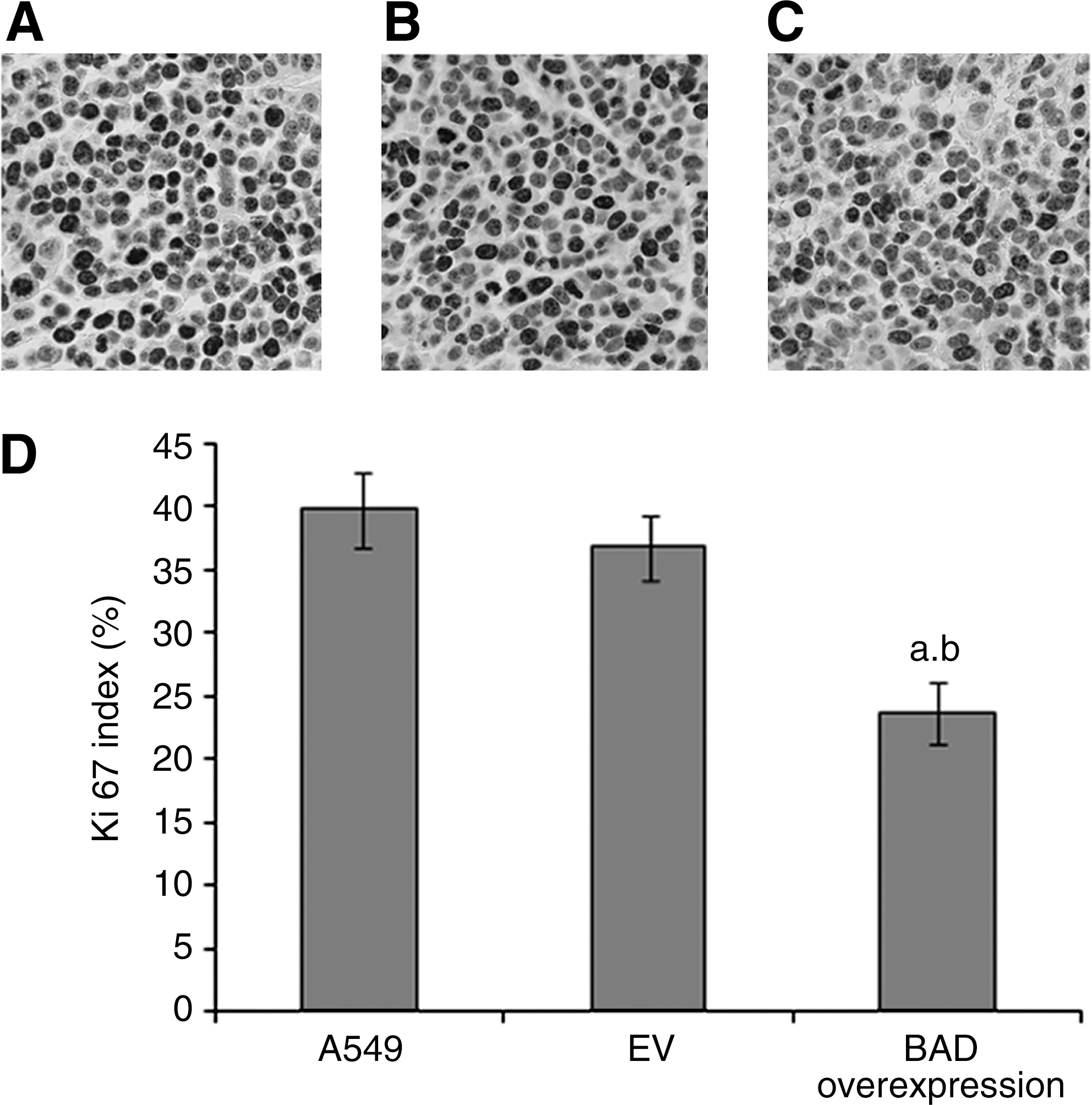
Representative tissue sections of immunohistochemical stained by Ki67.
To explore the role of BAD overexpression in apoptosis of A549 cells in vivo, apoptotic cells were visualized in situ through labeling of fragmented DNA in histological tissue sections of lung tumor xenograft by TUNEL. 22 The quantity of TUNEL-positive cells was higher in the BAD group than the EV and A549 groups (p<0.05) (Fig. 4).
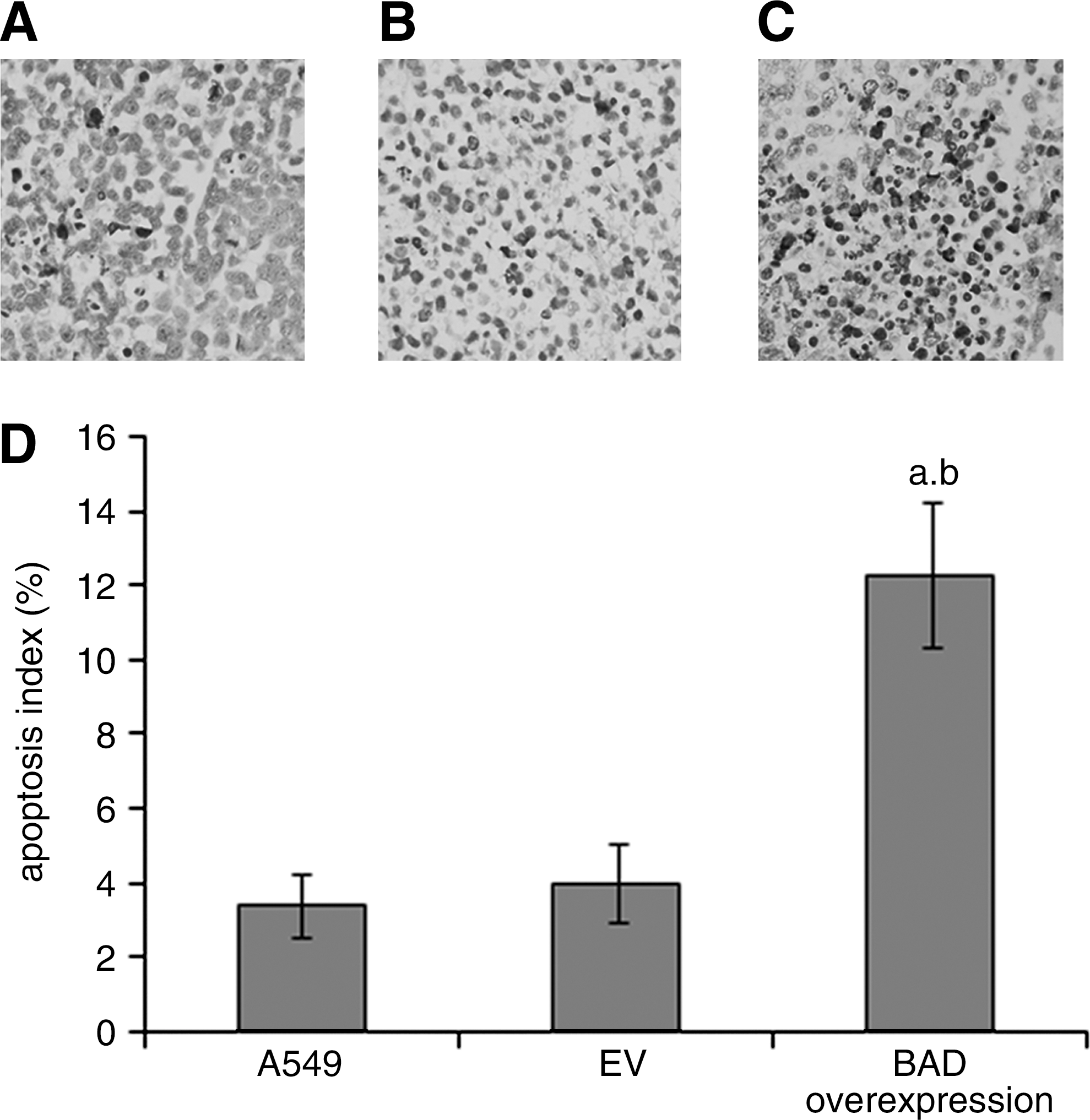
Representative tissue sections of formalin-fixed tumors stained by TUNEL.
Discussion
The results of our current study indicate that the overexpression of BAD inhibits lung cancer growth. Compared with the A549 and EV groups, the BAD group led to a 51.02% and 43.03% reduction of lung tumor volume, respectively. This maybe due to BAD overexpression that suppresses proliferation and promotes apoptosis of lung cancer cells, which has been verified in vitro and in vivo.
Growing interest is currently focused on the role of BAD in the regulation of cell survival and death. In our study, overexpression of BAD reduced lung cancer cell proliferation, which is consistent with the previous study. 23 Fernando et al. demonstrated that BAD overexpression inhibited G1 to S phase transition and estrogen-induced cyclin D1 synthesis in breast cancer. On the other hand, inhibiting endogenous BAD apparently increased cell proliferation, DNA synthesis, and cyclin D1 protein expression. 21 However, in contrast to our study, Smith et al. found that overexpression of BAD played a positive effect on cell division in prostate cancer, and knockdown of BAD expression by shRNA inhibited tumor growth. 18 The reason for these disparities maybe explained due to cell line variability. Molecular regulation mechanism of cancer cells is quite complex; the proteins are regulated by various kinds of signals. Therefore, the same protein could exert different effect on different cancer cells.
BAD overexpression not only plays a negative role in A549 cell proliferation, but it also promotes apoptosis of these cells. The mechanism of apoptosis depends on the balance between antiapoptotic and proapoptotic factors, which are regulated by survival signals and apoptotic stimuli. It has since been established that BAD phosphorylation and dephosphorylation regulates the response of cells to apoptotic stimuli by inducing mitochondrial dysfunction. 11 The active BAD is mainly located at mitochondria and binds to Bcl-XL and Bcl-2 to neutralize their antiapoptotic function by inducing BAX homodimer formation. 8 Our observations find that overexpression of BAD promotes apoptosis in lung cancer cells and this agrees with previous studies that BAD overexpression led to a significant increase in apoptosis of 293 cells 10 and CHO-K1 cells. 9 On the other hand, a previous study has demonstrated that decreasing the expression of endogenous BAD with siRNA notably attenuated apoptosis induction in response to inhibition of both EGFR/MEK/MAPK and PI3K/Akt pathways. 24 In addition, BAD-deficient mice developed diffuse large B cell lymphoma. 25
In summary, our results indicate that overexpression of BAD inhibits the growth of A549 cell line in vitro and in vivo by inhibiting cell proliferation and inducing apoptosis. Failure to regulate apoptosis is a common feature in tumor pathogenesis. Thus, induction of apoptosis in cancer cells becomes a valuable pathway in conquering cancers. Such strategies can involve direct induction of proapoptotic molecules. 26 Further, a synthesized BAD-BH3 peptide has been demonstrated to induce apoptosis in leukemia cells and it has delayed myeloid leukemia growth in mice. 27 Therefore, it is tempting to speculate that BAD could also be a potential target for lung cancer therapy.
Footnotes
Acknowledgments
The authors thank Wen-tong Meng, Yong-gang Zhang, Chun Xiao, Qiao-rong Huang, and other colleagues for excellent technical assistance in Laboratory of Stem Cell Biology. This study was supported by National Basic Research Program of China (Grant No. 2007CB947802). We appreciate Prof. Zhou Xing (Department of Pathology and Molecular Medicine, McMaster University, Canada), Mao-Bin Meng (Department of Thoracic Oncology, Cancer Center, West China Hospital of Sichuan University, China), and Jun-Yi Zhang (Corpus Christi College, Cambridge, United Kingdom) for modifying the manuscript.
Disclosure Statement
None declared.