
Abstract
The field of stem cell biology continues to evolve by characterization of further types of stem cells and by exploring their therapeutic potential for experimental and clinical applications. Human mesenchymal stem cells (hMSCs) are one of the most promising candidates simply because of their easiness of both ex vivo expansion in culture dishes and genetic manipulation. Despite many extensive isolation and expansion studies, relatively little has been done with regard to hMSCs' therapeutic potential. Although clinical trials using hMSCs are underway, their use in cancer therapy still needs better understanding and in vivo supporting data. The homing ability of hMSCs was investigated by creating a human xenograft model by transplanting an ovarian cancer cell line into immunocompromised mice. Then, genetically engineered hMSC-telo1 cells were injected through the tail vein and the contribution and distribution of hMSCs to the tumor stroma were investigated by immunohistochemistry and PCR specific to the telomerase gene. Results show that exogenously administered hMSCs preferentially home, engraft, and proliferate at tumor sites and contribute to the population of stromal fibroblasts. In conclusion, this study provides support for the capacity of hMSCs to home to tumor site and serve as a delivery platform for chemotherapeutic agents.
Introduction
The human adult bone marrow is the main source for mesenchymal stem cells (hMSCs). These cells can differentiate into a variety of adult mesenchymal tissues including bone, cartilage, adipose, muscle, ligament, and tendon. 1 –6 So far, hMSCs have been isolated from different tissues and have been very intensely studied and described. 7 –14 Combining the fact that hMSCs can differentiate into various cell types in vitro with their ease of in vitro expansion, hMSCs seem to be a promising source for tissue repair and gene therapy. Further, hMSCs are easily transduced by a variety of vectors; they can be envisioned as vehicles for both short-term and long-term gene therapy. The success of future clinical applications includes an exhaustive understanding of the biology of the hMSCs and, more importantly, the biological consequences of isolation, expansion, and manipulation of the stem cell for therapeutic use. Recently, hMSCs have been closely evaluated for different therapeutic potentials. 15 –18 Possible therapeutic potentials of hMSCs have been investigated in diverse clinical disorders such as myocardial infarction, diabetes, hepatic failure, acute renal failure, and cancer. 19,20 Recent studies have shown the potential of hMSCs in the treatment of various complications of diabetes mellitus, such as nephropathy, neuropathy, and wounds. 21 Another therapeutic approach to various conditions is to use stem cells as cellular vehicles.
Despite the fact that sparse information of the in vivo behavior of hMSCs is available in comparison to the information on their in vitro characteristics, several studies have been performed using hMSCs. These studies were mainly based on site-directed and/or systemic administration of the cells and both delivery methods of hMSCs have shown their ability of engraftment in a number of tissues after injury. 22 –25 In earlier studies, cultured MSCs have been infused into patients to support bone marrow transplant for osteogenesis imperfecta and glycogen storage disease, wherein their therapeutic options are limited. 26 They have also been used in graft versus host disease and their role in the treatment of Crohn's disease is being explored. 27 Although the exact mechanism is not well understood, introduction of MSCs into the infarcted heart directly or through intravenous administration have resulted in improved recovery and prevented deleterious remodeling. 26
It has been shown that hMSCs may contribute to tumor stroma formation by promoting angiogenesis or by creating a niche to support cancer stem cells survival. 28 Thus, hMSCs having the potential to home to the tumor stroma allows them to be a promising tool for the delivery of anticancer drugs to the tumor microenvironment. This strategy has been shown to work by the observation of specific homing of intravenously administered hMSCs, engineered to produce interferon-β (IFN-β), to tumors, with subsequent tumor regression in a xenogenic mouse model. 29 This study showed that MSCs expressing IFN-β could inhibit the growth of tumor cells in vivo. The approach required integration of the hMSCs at the tumor site, because nontumor site integrated or systemic delivery of IFN-β did not have enough tumor-regressing effect. On the other hand, in a recent study, Seo et al. have demonstrated that injection of interleukin (IL)-12M-expressing MSCs directly into the tumor had the strongest antitumor effect compared with other injection routes such as intravenous or subcutaneous. In addition to the inhibition of solid tumor growth, the same study also demonstrated antimetastatic effects for MSCs/IL-12M when embedded in the Matrigel. 30
As can be seen from the various examples given above, gene therapy based on targeted delivery has a promising future, especially in cancer treatments, but at present, systemic delivery in gene therapy is complicated by numerous factors including immune reactions, nonspecific accumulation in normal tissues, and poor permeation. Therefore, it is a significant advantage to have engineered vectors that localize to the sites of tumors before being released from the vehicle. One solution is to use cell-based carriers that preferentially home to the cancer site using the biological properties of tumor formation, thus serving as a delivery platform for therapeutic agents. The present study further investigated the homing ability of hMSCs using telomerase-immortalized bone marrow-derived mesenchymal stem cells (hMSC-telo1), 31 which has overcome the replicative exhaustion problem, to test whether the hMSCs can preferentially engraft at the tumor site and further can serve as vehicles for local delivery of biological agents to the tumor.
Materials and Methods
Cell culture
hMSCs immortalized with the hTERT gene (hMSC-telo1)
31
were used to generate hMSC-telo1-NTR cells by stably transfecting hMSC-telo1 cells with a nitroreductase plasmid (pd2ntr-CMV).
32
Cells were grown in high-glucose (4.5 g/L) Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal calf serum (Invitrogen), 100 U/mL of penicillin and streptomycin (Invitrogen), and 2 mM of
In vivo study (animal study)
All mice (NOD SCID) were inoculated with 106 A2780 cells (human ovarian cancer cell line). After the tumors appeared, one group (19 mice) was tail vein injected with 106 hTERT immortalized hMSCs (hMSC-telo1-NTR) and then subjected to CB 1954 prodrug treatment (2 mg per mouse diluted in 10% acetone and 90% peanut oil and delivered i.p.; Sigma-Aldrich). The prodrug was given 3 days and again 10 days after injection of hMSCs. Another group of mice did not receive the hMSC-telo1-NTR cells but received the prodrug treatment (7 mice). The third group (4 mice) received hMSC-telo1-NTR cells but instead of prodrug the mice received cisplatin 3 mg/kg i.v. (1 mg/mL cisplatin; MEDA A/S), which is a common therapeutic agent for ovarian cancer. The last two groups were control groups; one group received neither cells nor injection (4 mice) and the other group only received inoculation of human ovarian cancer cells and prodrug treatments (7 mice), which showed absolutely no effect as expected. After sacrificing the mice, the tumor tissue including surrounding stroma was primarily secured for immunofluoresence and PCR studies. However, in a number of cases, tissue was also available for fixation in formaldehyde, embedding in paraffin, and subsequent immunohistological studies.
RNA extraction and RT-PCR analysis
RNA was isolated from cells and tumors using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from RNA using the First Strand cDNA Synthesis Kit for RT-PCR (AMV; Roche) according to the manufacturer's instructions. Ectopic expression of hTERT was analyzed using a vector-specific sense primer (5′-GGACCATCTCTAGACTGACG-3′) together with an ectoTERT-specific antisense primer (5′-GGAGCGCACGGCTCGGCAGC-3′). Betain (5 M; Sigma-Aldrich) has been included as an enhancing agent to increase the yield, specificity, and consistency of PCRs in the ectoTERT-specific PCRs. β-Actin primers (sense: 5′-CTGTGCTGTCCCTGTATGCC-3′; antisense: 5′- GTGGTGGTGAAGCTGTAGCC-3′) were used in parallel reactions as a control of the cDNA preparation. Further, β-actin primers were used in a PCR performed on RNA to verify that no DNA contamination was present in the RNA preparation. Amplification parameters for β-actin and ectoTERT were as follows: an initial denaturation step at 94°C for 7 minutes, 38 cycles of 95°C for 30 seconds, 56°C for 30 seconds, and 72°C for 1 minute, followed by a final extension step at 72°C for 10 minutes.
Immunohistochemistry
Immunofluoresence staining of frozen tissue sections with AS02 (human-specific fibroblast mouse monoclonal antibody)
Six (6)-micrometer-thick frozen sections were cut on a cryostat and placed on slides, fixed with 4% neutral buffered formalin for 5 minutes, and washed three times in PBS with 0.1% Triton X-100. The slides were then blocked with blocking reagent (0.5% skimmed milk with 0.3% Triton X-100 and 15 mM sodium azide). The primary antibody AS02 (cat. no. DIA120; Dianova, Inc.) was diluted 1:200 in blocking reagent, added onto the slides, and incubated at 4°C overnight. The slides were then washed with PBS with 0.05% Tween 20 for 2×5 minutes and incubated with the secondary antibody goat anti-mouse Alexa Fluor® 488 1:400 (cat. no. A-11001; Molecular Probes/Invitrogen) for 1 hour in the dark at room temperature.
Immunohistological staining with CD90
Four (4)-micrometer-thick sections were cut from the paraffin blocks, melted at 60°C for 30–60 minutes, and then dried overnight at 37°C. Slides were then deparaffinized in Tissue Tek Prisma (autostainer) through Tissue Clear and rehydrated through graded series of ethanol (99%-96%-70%) to water. The sections were then washed in demineralized water for 5 minutes and pretreated in citrate buffer (pH 6.0) in a microwave oven (10 minutes full power to reach boiling point, followed by 15 minutes boiling and 15 minutes in the buffer to cool at room temperature). After washing in Wash buffer (0.05 M Tris, 0.015 M NaCl, and 0.1% Tween 20), inactivation of endogenous peroxidase activity was carried out using peroxidase blocking (cat. no. K4007; Dako) for 5 minutes, followed by wash in demineralized water (5 minutes). Incubation with primary anti-CD90 antibody (cat. no. 2694-1; Epitomics) diluted 1:100 in antibody diluent DAKO Real™ (cat. no. S2022; Dako) overnight at 4°C was followed by wash with Wash buffer (2×5 minutes). Detection was performed by HRP-Envision (cat. no. K4011; Dako) in 30 minutes, followed by wash with Wash buffer (2×5 minutes). The HRP reaction (DAB) took place for 15 minutes and was finalized with a wash in demineralized water for 5 minutes. Enhancement of the DAB reaction was accomplished with 0.5% copper sulfate in TBS (10 minutes) and counterstaining was carried out with Mayers hematoxylin (1 minute). After wash in tap water, dehydration in graded ethanol (70%-96%-99%) was followed by clearing in xylene (2 minutes) and mounting in pertex.
Results
Investigation of the homing ability of the hMSC-telo1 cells was done by coculturing these cells with A2780 cells (4:1 ratio) in vitro. Then the cocultured cells were injected into nude mice as xenografts (10 mice). Over a time course of tumor formation, mice were sacrificed and tumors were examined for the contribution of hMSCs to the tumor stroma. The presence of hMSCs was immunohistochemically detected with the monoclonal antibody ASO2 that is specific for human fibroblasts and mesenchymal cells in a mouse background. In addition, PCR against the vector sequence used for hTERT-immortalization was applied to detect and determine the hMSC distribution (data not shown). These results demonstrate that hMSCs, when coinjected, could participate in the formation of tumor stroma. It has been previously shown that hMSCs-telo1 cells retain their stem cell characteristic and differentiate into tissues such as adipose and bone. 31 Then, for further investigation of the homing ability of hMSCs, A2780 cells were subcutaneously injected on the back of nude mice as xenografts. When tumors appeared, genetically engineered hMSC-telo1 cells were injected through the tail vein. A week later, the mice were sacrificed and tumors were examined for the contribution of hMSCs to the tumor stroma. Using ASO2 and CD90 antibodies, the hMSC-telo1 cells could be mainly demonstrated at the peripheral part of the tumor (Fig. 1A–D). Distribution of the injected hMSCs to other sites in the body was also investigated and a fraction of the cells could be detected in lung tissue. This finding is in accordance with previous results of Bentzon et al. 33 Thus, the present study results show that exogenously administered hMSCs preferentially home, engraft, and proliferate at tumor sites and contribute to the population of stromal fibroblasts.

Immunofluorescence demonstrating the genetically engineered human mesenchymal stem cells (hMSCs) localized at tumor site.
Further, to be able to exploit the ability of hMSCs to deliver tumor-selective gene therapy vectors efficiently and specifically to the target sites of tumor stroma, mice injected with telomerase-immortalized hMSCs (hMSC-telo1) stably transfected with a vector that contains the NTR suicide gene (pd2ntr-CMV; bacterial nitroreductase) were analyzed.
Then, tumor xenografts were established by injecting the human ovarian cancer cell line A2780 under the back skin of nude mice. Following tumor growth, telomerase-immortalized hMSC-NTR (hMSC-telo1-NTR) were administered by tail vein injection and the mice were subsequently subjected to prodrug treatment (CB 1954). The release of nitroreductase bioactivates the prodrug CB 1954 into an active cytotoxic alkalizing agent, and in cell culture, the treatment of hMSC-telo1-NTR cells with CB 1954 leads to cell death (data not shown). In 11 of 19 mice injected with hMSC-telo1-NTR, presence of the hMSC-telo1-NTR cells was verified by PCR and IHC at the tumor site (Fig. 2A and Table 1). As seen in Figure 2B, results also show that this pharmacological induction can result in reduction of tumor size.
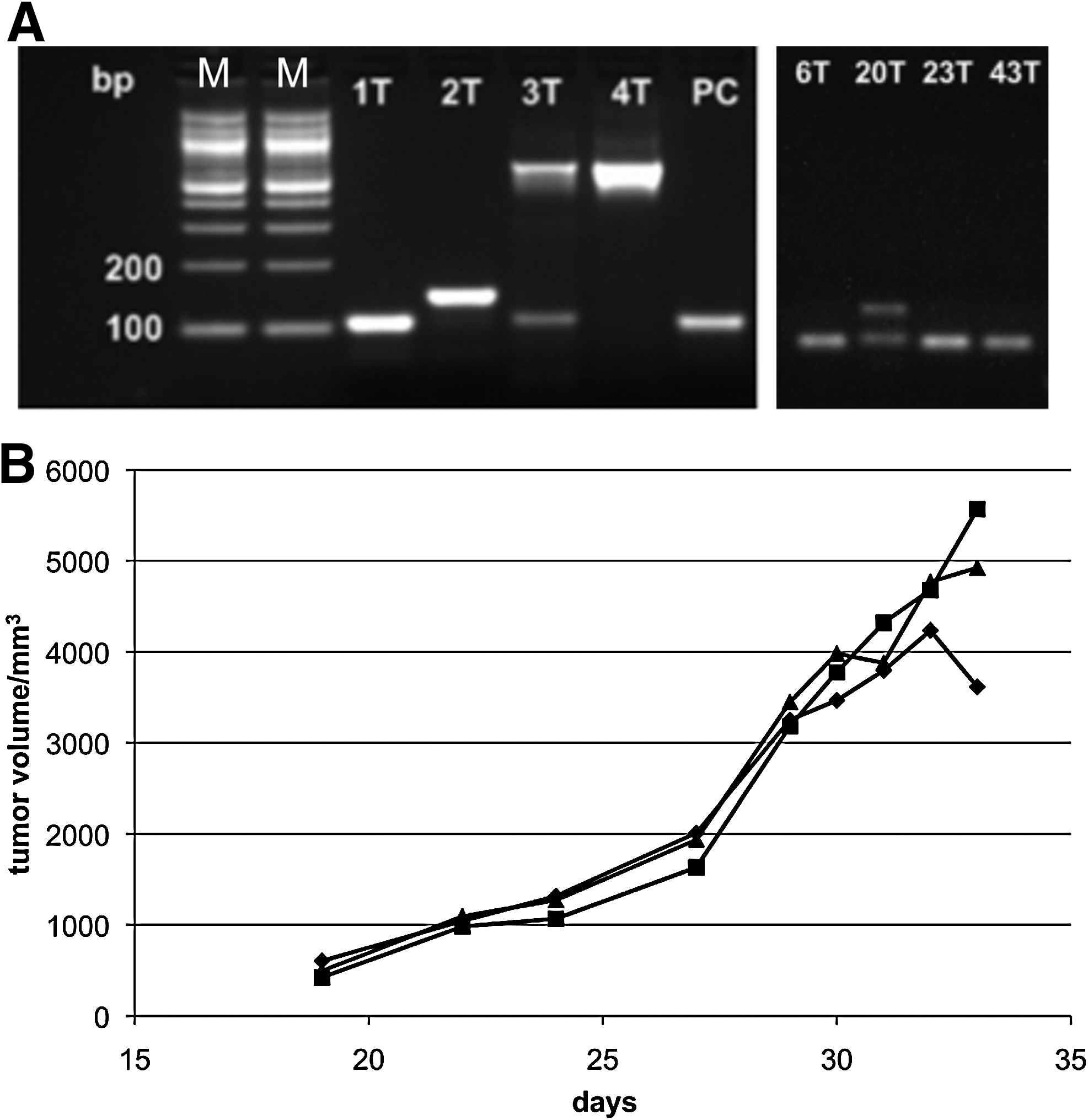
Analysis of selected tumors by PCR using primers recognizing vector sequences in hMSC-telo1-NTR cells (hTERT) and by immunohistological staining of CD90. GAPDH was used as a reference gene in the PCR analysis. (+) represents weak staining; n.d.: not determined as there was not enough tumor tissue for immunohistochemical evaluation.
Samples that gave a positive PCR result but was negative for CD90. In a number of cases there was not enough tissue available for immunohistological studies (necrosis and/or sampling error of the tumor periphery with surrounding tumor near stroma).
hMSCs, human mesenchymal stem cells.
The present study results therefore demonstrate both homing of the hMSCs at the tumor site (Figs. 1 and 2A) and occurrence of cellular targeting, which led to reduction of tumor size after the pharmacological induction (Fig. 2B).
Discussion
hMSCs can be easily obtained from bone marrow aspirates, expanded in vitro without loss of function, and genetically modified. For these obvious reasons, hMSC-based therapies provide a promising approach for the treatment of cancer. Moreover, hMSCs have been shown to home to some tissues, especially when injured or when some pathological conditions occurred. Previously, Barbash et al. 22 have demonstrated that systemic intravenous delivery of rat bone marrow MSCs to infarcted myocardium is feasible but still limited by the lung barrier. This study also showed that the site-directed administration of bone marrow MSCs directly to rat left ventricular cavity enhances migration and colonization of the cells preferentially to the ischemic myocardium. 22 Nonautologous MSCs that were intramuscularly injected can be safely used for the treatment of dystrophic muscle, by contributing to both the preexisting and new muscle fibers, without inflaming the immune system of the host. 34 The ability of MSCs to repair damaged tissue is thought to be primarily from their capacity to secrete paracrine mediators such as IL-10, IL-1ra, keratinocyte growth factor, and prostaglandin. 19 MSCs have also been suspected of migrating to the side of breast cancer and causing the tumor cells to be unresponsive to hormonal therapy. 35 As mentioned earlier, MSCs directly injected into sarcomas not only prevented tumor growth but also displayed a significant antimetastatic effect even in immonudeficient mice. 20 However, the mechanisms underlying migration/homing of hMSCs have yet to be clarified, even though there are some evidence suggesting the involvement of chemokines and their receptors. 36 –38 The following chemokine/receptor pairs have been implicated in MSC migration: SDF-1/CXCR4, SCF-c-Kit, HGF/c-Met, VEGF/VEGFR, and PDGF/PDGFr to name a few, in addition to cellular adhesion molecules. However, the migration to the tumor site is believed to be nonspecific, because the administered MSCs have been shown to localize to lung, bone marrow, and lymphoid organs. Additionally, it has been observed that whole-body radiation increases the distribution of MSCs to multiple organs. 20
Today, cancer remains as one of the most challenging diseases with regard to treatment. One of the biggest problems is that there is really no selective killing toward tumor cells. Thus, therapies that are more specifically directed toward cancer stem cells might result in much more durable responses and even might also cure the metastatic tumors. Based on their homing abilities, hMSCs have been used as cellular vehicles for local delivery of biological agents to brain tumors. Human MSCs were transduced with a lentivirus expressing secretable TRAIL (S-TRAIL) and mCherry (red fluorescent protein) and injected into established intracranial glioma tumors in mice. The genetically modified hMSCs were able to inhibit tumor growth, resulting in significantly longer animal survival. Thus, the study demonstrated the therapeutic efficacy of hMSC S-TRAIL cells and confirmed that hMSCs can serve as a powerful cell-based delivery vehicle for the site-specific release of therapeutic proteins. 39 In earlier experiments looking at targeted delivery, MSCs genetically engineered to secrete IFN-β were able to successfully home and engraft at melanomas growing in mice lungs and locally deliver IFN-β. When delivered locally, IFN-β was able to inhibit the growth of malignant cells both in vivo and in vitro. The same effect could not be achieved with systemically delivered IFN-β or IFN-β produced away from the tumor site. 29,40 In a recent experiment, MSCs were shown to be recruited to and incorporated into the prostate epithelium during prostate regrowth (after testosterone reintroduction). The incorporated MSCs were used to deliver frizzled related protein-2 (SFRP2) to antagonize the Wnt-mediated cancer progression by reducing tumor growth, increasing apoptosis, and causing potential tumor necrosis. 41
At this point, based on the aforementioned studies and the present study results, it can be concluded that using hMSCs as a vehicle has direct importance for therapeutic approaches to cancer. Thus, taken together, to date there are good indications that human bone marrow MSCs isolated from individuals and subsequently in vitro expanded can be used as delivery vehicles targeting tumor stroma. Therefore, delivery of therapeutic agents using MSCs to the tumor sites presents itself as a new therapeutic tool. The biggest advantage of using hMSCs as a therapeutic vehicle is that the tumor cells can be targeted while the normal cells are spared. Harnessing this targeted delivery potential of hMSCs will not only benefit cancer treatment but also have marked advantages for other systems where systemic delivery fails and targeted delivery becomes a necessity. However, there are some concerns regarding the clinical use of hMSCs. Considering that one of the hallmarks of cancer development is continued cell growth, which most often correlates with activation of telomerase, the question must be raised whether there is a potential risk of cancer from the genetically engineered hMSCs used for targeted cancer therapy. Wang et al. have noted that when MSCs were cultured, a subpopulation of cells showed high levels of telomerase activity, chromosome aneuploidy, and translocation and were capable of forming tumors in NOD/SCID mice. However, these results were not reproduced in subsequent studies. 20,42 It is important to also mention that there have been no reports regarding tumor seeding by MSCs in noncancerous tissues, suggesting the need for closer monitoring of hMSCs during therapy. 20 With regard to neoplastic potential of hMSCs, taking together the aforementioned controversial findings, caution is warranted but using hMSCs for targeted therapy should not to be discouraged. Suggestedly, to overcome the risk for neoplastic transformation of hMSCs in therapeutic use, different systems could be devised to address this during conduction of therapeutic approaches. Thus, it is critical to place a fail-safe system that will remove or inactivate the genetically engineered stem cells once they have homed to the tumor site and delivered the therapeutic agent. Momin et al. have thus suggested using MSCs from different sources, leading to a larger volume and therefore reaching sufficient numbers through few numbers of passages. Another approach might be to use telomerase inhibitors in an effort to stop the proliferation of the genetically modified MSCs. As the telomerase is essential to the life of these telomerased cells, treatment with telomerase inhibitors will eventually lead to cell death. However, this approach might raise yet another problem, namely, some of these cells might escape cell death, which will lead to genomic instability and ultimately cause the development of a new cancer. Alternatively, another and perhaps a safer approach might be to cause the vehicle cells to commit suicide after delivery of the targeted treatment. This strategy has been used in connection with tumor-selective genetically modified viruses that mediate oncolytic effects on tumors by replicating in and killing cancer cells in a targeted manner. These viruses contain transcriptional response elements based on the telomerase promoter sequence, thereby attacking telomerase-positive cells, which includes the genetically modified MSCs in this case. 32,43 –45
Conclusions
These studies have suggested that a therapy approach that combines genetically engineered vehicle stem cell therapy and the suicide gene therapy might improve targeting therapies and at the same time reduce the risk of secondary tumors. Still considering the lack of consensus regarding the neoplastic potential of hMSCs, that is, to be able to use hMSCs in targeted cancer therapy, a careful safety monitoring and preclinical tests are needed. In addition, improvements in culture strategies and better insights into the regulation of different cellular functions will help with the safety concerns and thus should be the focus of future investigations.
Author Contribution
N.S. conceived and designed the study, carried out the molecular genetic studies, coordinated the study, participated in cell culture work, and drafted the manuscript. R.C. carried out cell work. U.F. drafted the manuscript and helped in evaluating the data. F.B.S. carried out the immunohistochemistry studies. F.D.H. carried out the in vivo animal studies. M.H. participated in the molecular genetic studies. T.H.J. participated in the immunohistochemistry studies. S.K. and N.W.K. participated in design and helped draft the manuscript. All authors read and approved the final manuscript.
Footnotes
Acknowledgments
The work was supported by the Danish Medical Research Council (FSS) and European Union contracts FI6R-CT-2003-508842 (RISC-RAD) and LSHC-CT-2004-502943 (Mol Cancer Med).
Disclosure Statement
No financial conflicts exist.