
Abstract
Purpose:
Avidin-coupled monoclonal antibody MX35 (avidin-MX35) and astatine-211–labeled, biotinylated, succinylated poly-
Methods:
125I-avidin-MX35 and 211At-B-PLsuc were administered i.p. in nude mice. Tissue distributions were studied at various time points and mean absorbed doses were estimated from organ uptake of 211At-B-PLsuc. Studies of myelotoxicity were performed after administration of different activities of 211At-B-PLsuc.
Results:
We observed low blood content of both 125I-avidin-MX35 and 211At-B-PLsuc, indicating fast clearance. After sodium perchlorate blocking, the highest 211At uptake was found in kidneys. Red bone marrow (RBM) accumulated some 211At activity. Mean absorbed doses of special interest were 2.3 Gy/MBq for kidneys, 0.4 Gy/MBq for blood, and 0.9 Gy/MBq for RBM. An absorbed dose of 0.9 Gy to the RBM was found to be safe. These values suggested that RBM would be the key dose-limiting organ in the proposed pretargeting scheme, and that blood data alone was not sufficient for predicting its absorbed dose.
Conclusions:
To attain a favorable distribution of activity and avoid major toxicities, at least 1.0 MBq of 211At-B-PLsuc can be administered 24 hours after an i.p. injection of avidin-MX35. These results provide a basis for future i.p. therapy studies in mice of microscopic ovarian cancer.
Introduction
In radioimmunotherapy (RIT) antibodies are labeled with radionuclides for specific targeting of tumors. When injected into a patient, these radioimmunoconjugates irradiate tumor cells as the attached nuclides decay. The effectiveness of the therapy depends to a great extent on the choice of radionuclide, and it is the intended target that determines what kind of radiation is most appropriate. When treating residual microscopic tumors, α-emitters have many advantages over the more commonly used β-emitters. Their short particle range in tissue (40–100 μm) theoretically enhances the specificity of the therapy, and the high linear energy transfer generates large numbers of irreparable DNA double-strand breaks even with a few α-particle passages. 1 –3 Among the available α-emitters, the cyclotron-produced nuclide astatine-211 (211At, t ½=7.2 hours) is one of the most promising.
Ovarian cancer spreads by forming micrometastases in the abdominal cavity, and intraperitoneal (i.p.) α-RIT is expected to be an effective treatment method. This has been confirmed by RIT studies with 211At-labeled MX35 IgGs or F(ab′)2 fragments in mice. 4,5 A phase I study of i.p. 211At-RIT in ovarian cancer patients was recently conducted at the Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden. 6 However, preclinical studies showed that 211At-RIT was mainly efficient at eradicating tumors up to diameters of 0.1–0.2 mm. 7 This might be due to restricted or slow penetration of labeled antibodies into the tumors. Theoretically, a smaller molecule should be able to penetrate tumors more efficiently. In pretargeted radioimmunotherapy (PRIT), the administration is separated into a targeting step (pretargeting molecule) and an activation step (effector molecule). 8,9 The effector molecule is much smaller than the pretargeting molecule, which facilitates its penetration into the tumor. It is also designed to quickly bind to the cell surface-bound pretargeting molecule, thereby yielding high uptake of activity in a short time compared with RIT. We believe that these factors might improve the therapy of ovarian cancer even further, using PRIT. Several PRIT regimens have been evaluated in clinical studies over the years, and they have shown good potential. 10 –14 Yet, to our knowledge, no such studies have been performed in an i.p. setting.
We previously reported promising in vitro results with a pretargeting system that consisted of an avidin-conjugated monoclonal antibody (MAb) and a polylysine-based, biotinylated, 211At-labeled, succinylated effector molecule.
15
Poly-
The aim of the current study was to evaluate this pretargeting system in an in vivo mouse model, prior to therapy studies. We used a small polymer base (3865 Da) for the effector molecule, and for the pretargeting molecule, avidin was conjugated to the MAb MX35 that specifically binds to an ovarian cancer cell line. The use of avidin instead of streptavidin in the conjugation of MX35 eliminates the need for a clearing step in future i.p. therapy studies, as avidin rapidly directs the unbound antibody from the circulation to the liver. The main objectives were to determine a timeline for administration, identify dose-limiting organs, and estimate the maximum tolerable activity (MTA).
Materials and Methods
General
Poly-
The MAb MX35 recognizes a membrane transporter molecule (NaPi2b) that is homogeneously expressed in about 90% of human ovarian epithelial cancers. 19 The human ovarian cancer cell line NIH:OVCAR-3 was obtained from the American Type Culture Collection and cultured at the Department of Oncology at Sahlgrenska University Hospital.
The 211At was produced via the 209Bi(α,2n)211At reaction at the Cyclotron and PET Unit, Rigshospitalet, and transported to the Department of Nuclear Medicine at Sahlgrenska University Hospital. The astatine in the irradiated target was isolated by dry distillation as previously described. 20 No-carrier added [125I]NaI was purchased from Perkin Elmer Sverige AB. Radioactive samples with high activity (>100 kBq) were measured in an ionization chamber (Capintec; CRC-15 dose calibrator), and low-activity samples (<10 kBq) were quantified with a NaI(Tl) γ-counter (Wizard 1480; Wallac).
Synthesis of the effector molecule, 211At/125I-B-PLsuc
Biotinylation of poly-l -lysine
NHS-dPEG®
4-Biotinidase-resistant biotin was dissolved in dimethylformamide to 6 mg/mL and immediately added at twofold molar excess to poly-
Conjugation with m-MeATE
N-succinimidyl-3-(trimethylstannyl)benzoate (m-MeATE; Toronto Research Chemicals, Inc.) was dissolved in chloroform at a concentration of 50 mg/mL. From this stock solution, 2 μL was removed, placed in a glass vial, and the chloroform was evaporated. The dry residue was redissolved in 15 μL dimethyl sulfoxide to a concentration of 17 mM. Two molar equivalents (13.7 μL) of m-MeATE were added to 0.45 mg of biotinylated poly-
Labeling with 211At
After the astatine was dry distilled, the radionuclide remained dissolved in chloroform, which was evaporated under gentle nitrogen flow. To oxidize the astatine (34.8–68.6 MBq) into a reactive form, 15–20 μL of N-iodosuccinimide (NIS) in methanol/1% acetic acid (130 μM) was added; immediately afterward, 200–350 μg of the succinylated m-MeATE-biotin-poly-
Labeling with 125I
Iodination of the m-MeATE-biotin-poly-
Synthesis of the pretargeting molecule, avidin-MX35
Conjugation
The pretargeting molecule was synthesized by conjugating avidin to the antibody, as previously described. 15 Briefly, avidin was activated by reaction with 4-(N-maleimidomethyl)cyclohexane-1-carboxylic acid 3-sulfo-N-hydroxysuccinimide ester sodium salt (sulfo-SMCC), and MX35 was thiolated by reaction with 2-iminothiolane hydrochloride. Equimolar amounts of thiolated MX35 and sulfo-SMCC–activated avidin were mixed and incubated. The resulting avidin-antibody monomers were isolated by size-exclusion fast protein liquid chromatography, and nonavidin-bound conjugates were removed by iminobiotin affinity chromatography. The buffer solution was exchanged to PBS for storage, or 0.2 M carbonate buffer (pH 8.5) for conjugation with m-MeATE and subsequent radiolabeling.
Labeling with 125I
For in vivo distribution detection the avidin-antibody conjugate was labeled with 125I. The iodination was performed according to the protocol for the effector molecule, with a few adjustments. In short, 125I (3.5 MBq) was activated by the addition of 5 μL of NBS in methanol/1% acetic acid (130 μM); then, 700 μg of m-MeATE–conjugated avidin-MX35 in 0.2 M sodium acetate buffer (pH 5.5, 2 mg/mL) was added to the reaction vial. After 1 minute of incubation, 15 μL of NIS in methanol/1% acetic acid (18 mM) was added, and the mixture was incubated for 1 minute. The 125I-avidin-MX35 product was subsequently isolated on a Sephadex NAP-5-column and eluted in 0.9 mL PBS.
In vitro analysis
The degree of biotinylation and the number of free amino groups on the m-MeATE-biotin-poly-
The quality of the labeled products was assessed in terms of radiochemical purity (RCP). The RCP was evaluated by trichloroacetic acid precipitation for 125I-labeling and instant thin-layer chromatography (ITLC) for 211At-labeling, in addition to size-exclusion chromatography. To ensure that the binding of the effector molecule to pretargeted cancer cells was unimpaired by labeling, in vitro pretargeting was carried out with a previously described cell assay. 15
To assess the bond stability between avidin-MX35 conjugates and cancer cells over time, a cell assay was performed with 125I-labeled biotin-poly-
In vivo procedures
All animal experiments were conducted according to protocols approved by the ethics committee of the University of Gothenburg. The tissue distributions of the iodinated pretargeting molecules and the astatinated effector molecules were evaluated in athymic mice. The mice (Balb/C nu/nu) were 5–9 weeks of age, and were fed a biotin-deficient diet (Harlan Teklad) for 5–7 days before the experiments. For measuring the distribution of 125I-labeled avidin-MX35, 25 mice were administered the iodinated pretargeting molecule (40 kBq/25 μg in 750 μL of PBS, i.p.). At 1, 3, 6, 10, and 24 hours postinjection (p.i.), the animals were sacrificed by cervical dislocation (n=5 animals per time point). Blood was collected immediately by cardiac puncture. Tissues (heart, lungs, salivary glands, throat, stomach, liver, spleen, kidneys, muscle, small intestine, large intestine, and i.p. fat) were removed and weighed, and their radioactive content was measured with corrections for decay and background.
To determine the distribution of the effector molecule, 211At-B-PLsuc (500 kBq/5 μg in 750 μL of PBS) was injected i.p. After 1, 3, 6, 10–11, and 24 hours, animals were sacrificed (n=5 animals per time point). Blood and tissues were collected as described previously. To reduce unspecific uptake of free astatine, a subsequent study was performed according to the same protocol, except that sodium perchlorate (NaClO4·xH2O; Sigma-Aldrich Sweden AB; 1.2 μmol/g in 100 μL of PBS) was administered i.p. twice prior to the injection of the 211At-labeled effector molecule, once at −24 hours and again at −1 hour, as proposed by Larsen et al. 23 At 6 hours after injection, 5 animals that had not been given any blocking agent before receiving the effector molecule were also sacrificed to serve as a control group.
Supplementary studies were also performed to evaluate the uptake of 211At-B-PLsuc in the kidneys at early time points, and to assess the uptake in red bone marrow (RBM). A biotin-deficient diet and blocking agent were provided as described previously, and animals were injected with 211At-B-PLsuc (500 kBq/5 μg in 750 μL of PBS, i.p.). At 15 minutes, 30 minutes, and 3 hours p.i., animals were sacrificed (n=5 animals per time point). The blood, kidneys, and RBM were harvested and analyzed. In another study, 211At-B-PLsuc was injected and at 1, 3, 6, 9, and 22 hours, only blood and RBM was collected (n=4 animals per time point). In that study, to ensure sufficient activity in the samples from the last time point, the 22-hour group was given 800 kBq/8 μg in 750 μL PBS, and all other animals received 500 kBq/5 μg as previously described. The bone marrow was collected from the femoral bone by cutting off the epiphysial ends and pushing out the marrow with a thin needle. To ensure accuracy in this procedure, only bone marrow samples of >0.5 mg were analyzed. The last study was later repeated, using 600 kBq/5 μg in 750 μL PBS for all animals and time points.
Myelotoxicity was evaluated by measuring the white blood cell (WBC) suppression after administration of the effector molecule. A total of 25 animals were provided with biotin-deficient diet and blocking agent as described previously. The mice were injected i.p. with 211At-B-PLsuc (0.24 MBq/4 μg; 0.60 MBq/5 μg; 0.77 MBq/5 μg; 1.04 MBq/5 μg in 750 μL of PBS) or B-PLsuc (5 μg in 750 μL of PBS), and the WBCs were counted at days 0, 5, and 14 p.i. (n=5 animals per group).
Finally, a small study including 4 tumor-carrying mice was set up to confirm uptake of the effector molecule in pretargeted tumors using the α-camera technique, a new method for quantitative ex vivo autoradiography in which the intensity signal on the resulting images is proportional to the activity level. 24 The animals (5 weeks of age) were inoculated i.p. with 1×107 NIH:OVCAR-3 cells in 0.2 mL of cell medium, which were allowed to grow for 7 weeks. Prior studies of this tumor model have shown small tumor clusters growing on the peritoneal surface and that a typical location was around the spleen. 7 For uptake studies, the animals were injected i.p. with either 211At-labeled MX35 F(ab′)2 (6.4 MBq/20 μg in 750 μL of PBS; n=2) or 211At-B-PLsuc (6.5 MBq/1 μg in 750 μL of PBS; n=2). The high activities were given to ensure good image quality. In the latter case, avidin-MX35 (25 μg in 750 μL of PBS) had been administered i.p. 24 hours before the effector molecule. All animals were provided with biotin-deficient diet and blocking agent as previously described. At 4 hours p.i. the animals were sacrificed and their spleens removed and immediately frozen and prepared for cryosectioning. Couples of 12-μm-thick sections at different levels of the spleen were cut using a cryostat microtome (HM 520; Microm International GmbH). For each pair of consecutive sections, the first section was used for α-camera imaging and the second section was H&E stained and used for morphological identification. After digitalization, the H&E sections were used for overlay with the α-camera images using the Photoshop CS5 software (Adobe Systems, Incorporated), and uptake in tumor cluster areas and in the spleen could be quantified. Small regions of interest (ROIs; diameter 48 μm) were drawn on the α-camera images and the mean pixel value was derived using ImageJ software (National Institutes of Health). The mean pixel intensities in tumor cluster areas from 211At-MX35 F(ab′)2 and 211At-B-PLsuc were compared with each other, as well as with spleen activity uptake derived from the same images.
Dosimetry calculations
The data collected from animal experiments were processed to express the percent of injected activity per gram of tissue (%IA/g). These were then used to estimate the mean absorbed doses. In this study, only the contribution from α-particles was taken into account, and the absorbed doses to various organs and tissues were calculated as follows:
Results
Chemistry
The 211At-labeling of the m-MeATE–conjugated, biotinylated polymer was efficient, with mean radiochemical yields of 84.8%±3.2% (SD). The RCP of the labeled product was 98.9%±1.0% (SD) as determined by ITLC, and fast protein size-exclusion chromatography confirmed that the quality was good. For 125I-labeling, the mean radiochemical yield was 88.0%±12.9% (SD) and the RCP was 99.9%±0.2% (SD). The biotinylation efficiency, determined by the HABA method, was 75.7%±1.5% (SD), meaning that for each 2 mol of biotin reagent added to the reaction mix, around 1.5 were bound to the polymer. The TNBSA analysis concluded that the final product contained essentially no free amino groups from the poly-
The avidin-antibody conjugation rendered an overall yield of 11.6%±5.5% (SD), determined by dividing the amount of isolated monomeric avidin-MX35 by the total MX35 added initially.
The results from the cell assay used to evaluate the bond stability between avidin-MX35 conjugates and OVCAR-3 cells in suspension showed that the binding of 125I-B-PLsuc decreased somewhat from 1 to 6 hours. The bound fraction (bound activity divided by total administered activity) after 6 hours of incubation was 64% of the bound fraction after 1 hour of incubation. After 6 hours, the decline was more gradual, with bound fractions after 24 and 48 hours of incubations of 62% and 57% of the 1-hour value, respectively (data not shown).
In vivo distribution
In the first animal study, the kinetics of the pretargeting and effector molecules were evaluated. The distributions among selected organs and tissues are shown in Figures 1 and 2. We expected that the glycosylated avidin would direct the antibody conjugate promptly to the liver, where it would be degraded. Indeed, the distribution shows that this was the case (Fig. 1). The radioactivity was rapidly cleared from blood, and the liver uptake reached a maximum after 3 hours. Twenty-four (24) hours after injection, the %IA/g was low in all tissues.
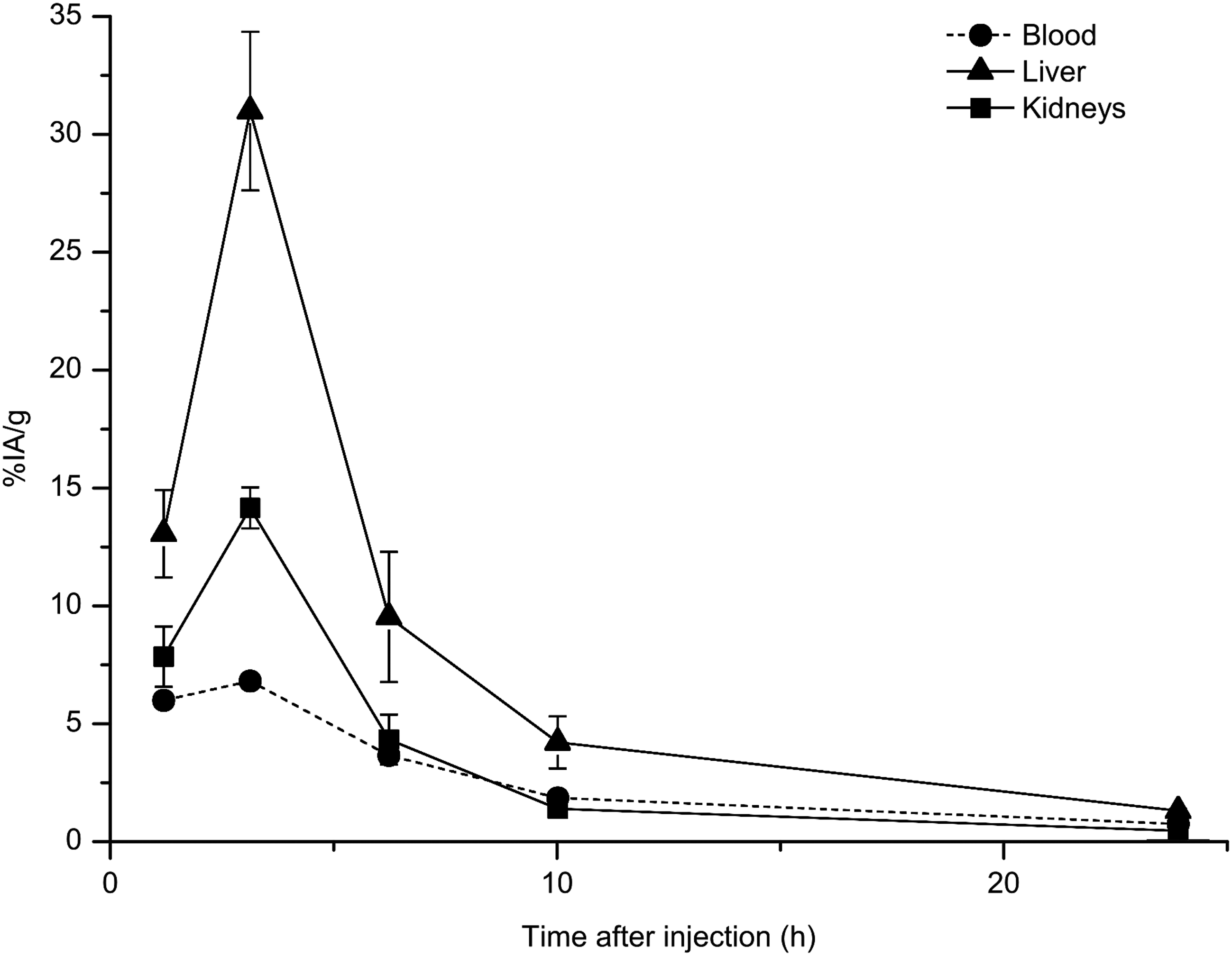
Distribution of 125I-avidin-MX35 after intraperitoneal (i.p.) administration in tumor-free mice. For blood, the median value is shown; for liver and kidney uptake, the mean±standard error of the mean (SEM) is shown. n=5 at each time point.

Distribution of 211At-B-PLsuc after i.p. administration in tumor-free mice. The uptake after 1–24 hours is expressed as mean±SEM, except that blood is expressed as the median value for each time point. n=5 animals per time point.
Due to the i.p. injection route, the blood concentrations of activity that derived from the effector molecule generally never reached high values, because the polymer-based molecule was quickly cleared from the circulation (Fig. 2). Because of large scatter in the blood data, we have chosen to report the median value for blood consistently in these results; in contrast, all other values are reported as the mean value±standard error of the mean.
Tissues that are known to accumulate astatine, including throat, salivary glands, lungs, and stomach, showed some uptake after injection of 211At-B-PLsuc. Presumably, this could have been reduced by administrating a suitable blocking agent. The effect of blocking astatine uptake was examined in a subsequent study, where sodium perchlorate was administered twice before the injection of the effector molecule. The results are shown in Figure 3; the most significant outcome was that the uptake in throat was reduced by almost ninefold. The uptake in the stomach and salivary glands was reduced by approximately five- and threefold, respectively. In lungs, the effect was more modest with a decrease that was less than twofold.
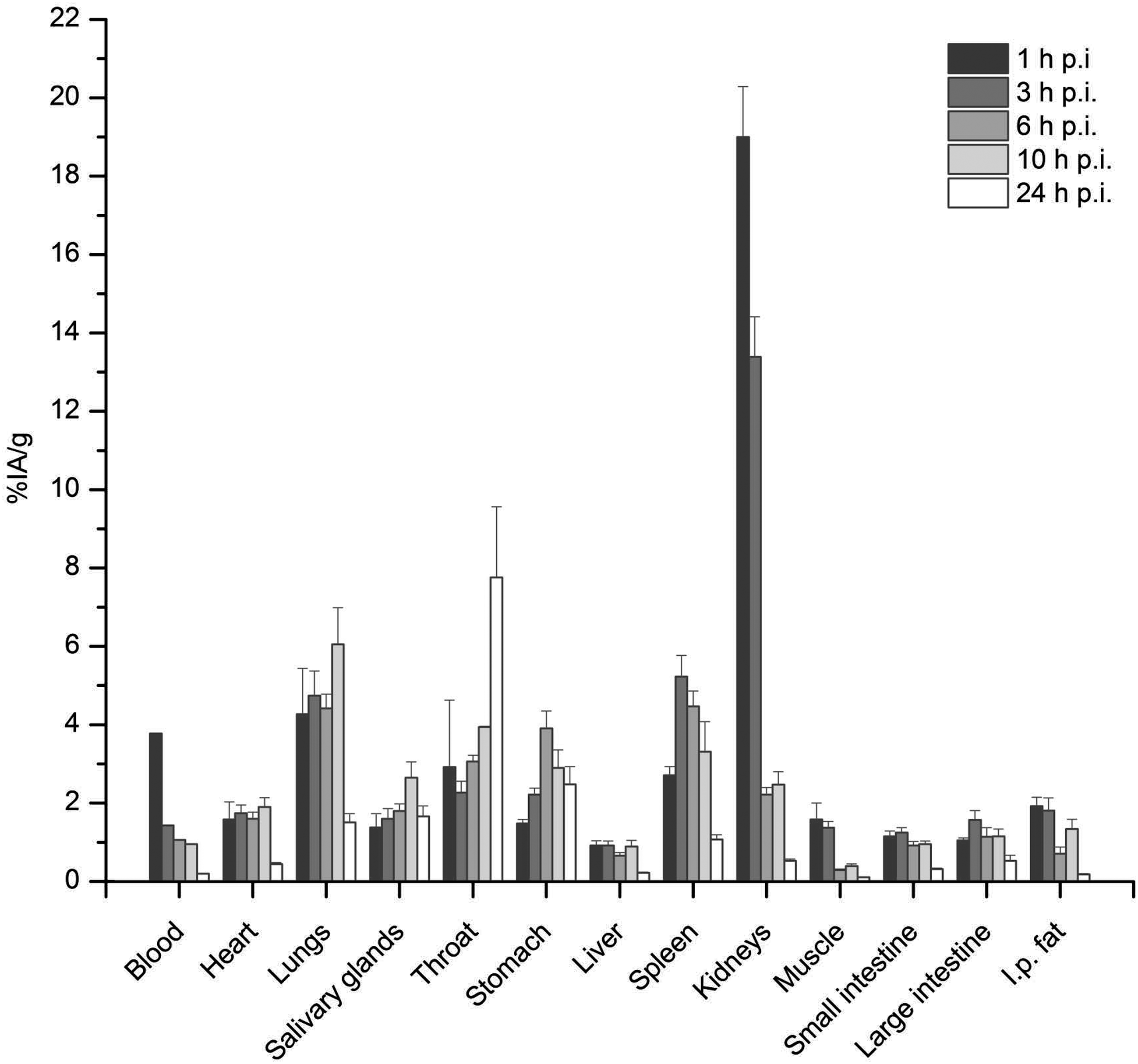
Distribution of 211At-B-PLsuc after i.p. administration in tumor-free mice. To block uptake of free astatine, the animals were given sodium perchlorate at 24 hours and 1 hour before injection of the astatine-labeled molecule. The uptakes at 1–24 hours after astatine injection are expressed as the mean±SEM, except the values for blood, which are represented by the median value at each time point. n=5 animals per time point.
The uptake of 211At-B-PLsuc in liver and kidneys indicated that this small molecule was cleared mainly by renal filtration. At first, the uptake in kidneys was studied from 1 hour to 24 hours p.i.; however, because clearance was rapid, we also studied earlier time points (15 and 30 minutes p.i.). The combined data in Figure 4 revealed that the peak uptake occurred at around 1 hour p.i., with an uptake of 19%IA/g. After that, uptake decreased rapidly and at 6 hours p.i., only about 2%IA/g remained in the kidneys.
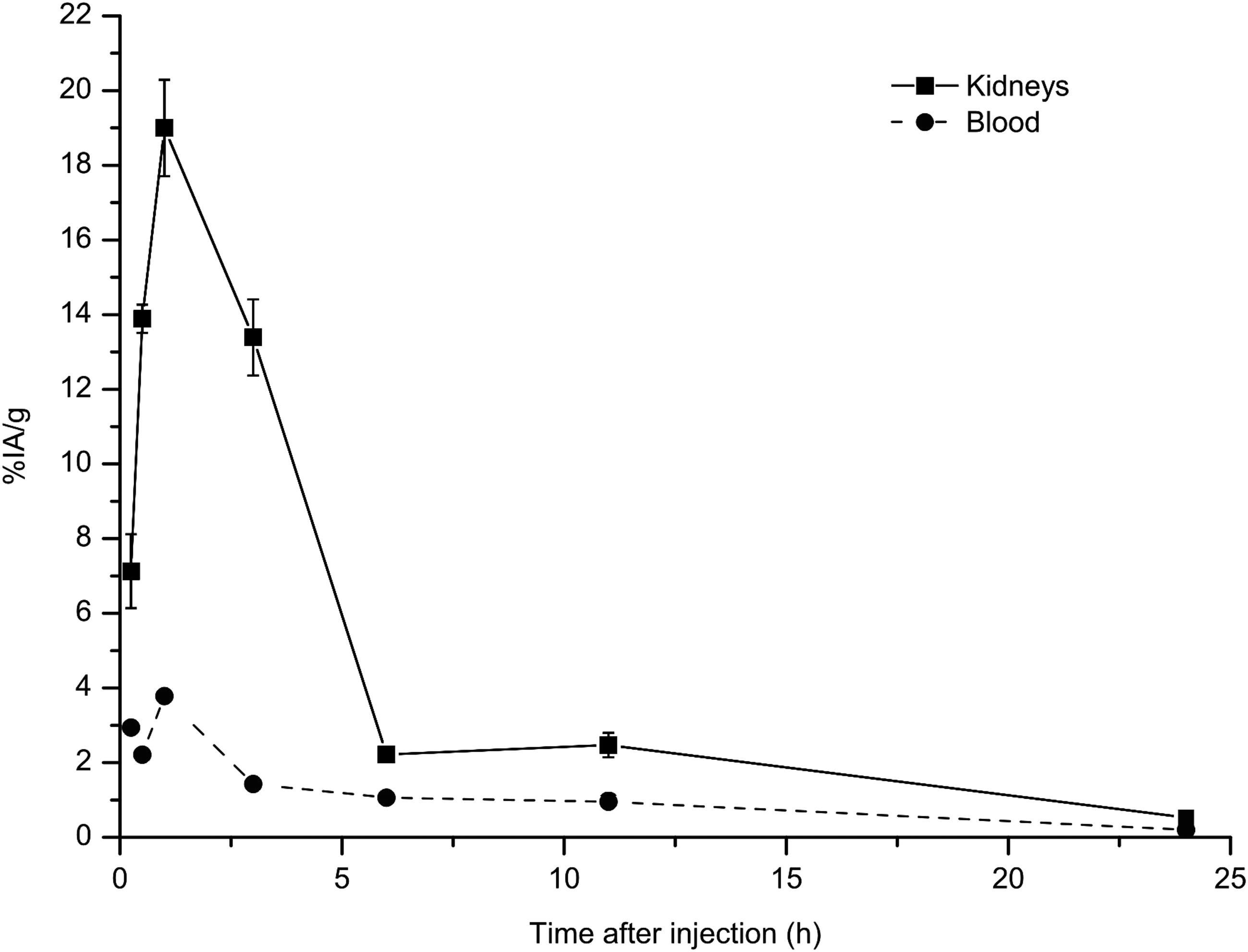
Combined results from distributions of 211At-B-PLsuc in tumor-free mice. Data from 1 hour to 24 hours after injection are combined with uptake at 15 and 30 minutes after injection from an additional study. To block uptake of free astatine, all animals were given sodium perchlorate at 24 hours and 1 hour before injection of the astatine-labeled molecule. The blood uptake is represented by the median value for each time point, but uptake in the kidneys is expressed as the mean±SEM. n=5 animals per time point.
The uptake of 211At in RBM indicated that its activity concentration was not directly correlated to the activity concentration in blood. Figure 5 shows that the bone marrow-to-blood ratio (BMBLR) increased steadily until at least 22 hours p.i.
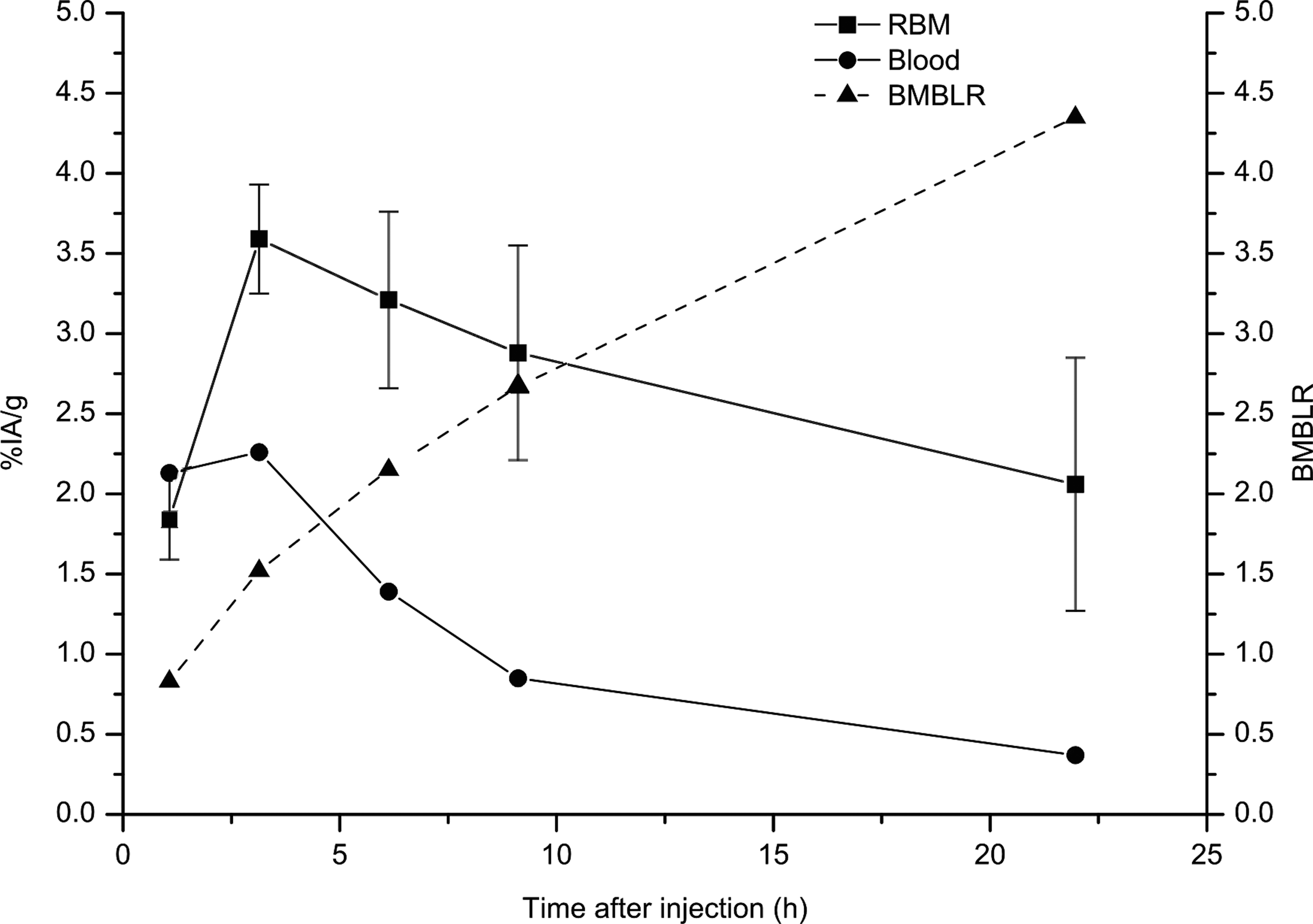
Uptake of 211At after i.p. injection of 211At-B-PLsuc in tumor-free mice, combined from two separate studies. Blood values are presented as median values; red bone marrow (RBM) uptake is presented as the mean±SEM. The bone marrow-to-blood ratio was calculated as the activity in RBM divided by the activity in blood at each time point. To block uptake of free astatine, the animals were given sodium perchlorate at 24 hours and 1 hour before injection of the astatine-labeled molecule. n=4 animals per time point for each of the two studies.
Estimations of mean absorbed doses in Gy per injected MBq of 211At-B-PLsuc are shown in Table 1. For kidneys, the estimated mean absorbed dose was 2.3 Gy/MBq based on combined data from the two separate experiments shown in Figure 4. For RBM, the mean absorbed dose was 0.9 Gy/MBq estimated from combined results of two separate studies. The administered activities in the myelotoxicity study consequently corresponded to doses of 0.0, 0.2, 0.5, 0.7, and 0.9 Gy. No severe toxicities were observed in any of the dose groups. Compared with the control group (0.0 Gy), the highest WBC suppression was approximately 75% at 5 days p.i. for 0.7 and 0.9 Gy. Averaging the individual values for each dose group yielded a maximum suppression to 36% at 5 days p.i. for 0.9 Gy. At 14 days p.i., all animals had recuperated their WBC counts, compared with their initial value at day 0 (data not shown).
To block uptake of free astatine, the animals were given sodium perchlorate at 24 hours and 1 hour before injection of the astatine-labeled molecule.
n=5 for each time point, except RBM, where n=4.
Calculated from the uptake in throat and a standard weight of thyroid of 3 mg.
Summarized from two separate experiments.
i.p., intraperitoneal; RBM, red bone marrow.
The resulting images from the comparative in vivo study of tumor uptake are shown in Figure 6. There is an evident uptake of 211At-B-PLsuc in tumor cluster areas compared with the uptake in spleen (Fig. 6A), which is similar to that of 211At-MX35 F(ab′)2 (Fig. 6B), which proves that the pretargeting system works as anticipated. For the pretargeted case, the mean pixel intensity per area unit is approximately five times higher for tumor than for spleen. The overlay images in Figure 6C and D show that the high activity areas correspond to clusters of tumor cells surrounding the spleen.

Alpha camera images of cryosections of the spleen showing the activity distribution of 211At-B-PLsuc
Discussion
Pretargeting methods might improve the potential of 211At-RIT for the treatment of disseminated ovarian microtumors. It could provide better penetration of labeled substances into tumors, and may also improve the therapeutic index of the treatment; that is, reduce irradiation of normal tissues and maintain a high dose to tumors. Promising results from a number of research groups have shown that PRIT may be superior to RIT, regarding efficacy and toxicity for hematologic and other malignancies. 25,26
The objective of the current study was to determine some basic characteristics of the proposed pretargeting system in an in vivo mouse model, prior to therapy studies. We aimed to gain information on the dosing and timing for administering avidin-MAbs and 211At-labeled, biotinylated, succinylated poly-
Previous in vitro results from pretargeting with avidin-MAbs and 211At-labeled, biotinylated, succinylated poly-
Before testing the pretargeting system in therapy studies, organs at risk for toxicity should be identified, and a time scheme for the injections should be designed. The timing should maximize the therapeutic effect and limit the doses to organs and tissues at risk. Thus, we performed in vivo distributions to assess the uptake in normal tissues of interest. This provided information for dosimetric calculations and estimations of the MTA. The distribution data also provided fundamental information regarding the optimal time between administrations of pretargeting and effector molecules.
Kidneys are generally considered a possible risk organ in RIT, and Bäck et al. 27 have studied the effect on glomerular filtration rate (GFR) in nude mice after α-RIT with 211At-labeled MX35-F(ab′)2 fragments. They concluded that a mean absorbed dose of about 10 Gy rendered a decrease in GFR of approximately 50%, which was considered a tolerable effect. The renal uptake of 211At reported herein resulted in an absorbed dose to the kidneys of 2.3 Gy/MBq. This suggested that the kidneys were unlikely to be dose limiting for our effector molecule.
RBM is very sensitive to radiation, and thus, it is more likely to limit the administered activity. 28 The dose to RBM is generally approximated from the absorbed dose to blood by estimating the BMBLR, which is assumed to be static. In this case clearance was rapid, which led to a low absorbed dose to the blood. However, we also studied the activity content in RBM; there, we discovered an unforeseen accumulation of activity that rendered an increase in BMBLR over time. This must be taken into account when estimating the MTA, and it precludes the use of blood data alone. We do not know whether the astatine is dispersed in a free or polymer-bound form in the RBM, or where it is situated in the marrow, but in our study evaluating the effect on bone marrow after administration of 211At-B-PLsuc, the highest absorbed dose (0.9 Gy), suppressed the WBC counts by at most 75%. The animals are believed to be able to recuperate from a WBC suppression of 90%, which means that 0.9 Gy can be considered a safe absorbed dose in this case. The estimated mean absorbed dose to the RBM implied that at least 1.0 MBq of the 211At-B-PLsuc-conjugate could be administered without severe toxicities, which is an activity that has proven curative for RIT with 211At-labeled antibodies. 4
We successfully used sodium perchlorate as a blocking substance to reduce unspecific uptake of 211At into organs that are known to accumulate free astatine. When comparing the biodistributions resulting from the unblocked and blocked schemes, it is evident that free 211At was released following the rapid metabolism in the kidneys. However, after administration of sodium perchlorate, the %IA/g was drastically diminished, yielding overall low uptakes in the analyzed tissues. Still, there is room for some improvement. The administration scheme used in this study was proposed by Larsen et al., 23 who also showed that sodium thiocyanate could further decrease the uptake in lungs and spleen. They suggested that multiple substances might block most efficiently. We believe that perchlorate and thiocyanate in concert may be a feasible way to minimize the accumulation of 211At in normal tissues.
For the timeline of the injections, the main consideration is the clearance of pretargeting molecules from the peritoneal cavity. Any residual avidin-MAb-conjugates would swiftly eliminate the biotinylated effector molecule; this would hinder it from reaching the pretargeted tumor cells, and thus, would drastically reduce tumor uptake. When analyzing the distribution of the iodinated pretargeting molecule, it is important to note that 125I, not the pretargeting molecule, is detected; thus, uptake in iodine-accumulating tissues, like the thyroid and stomach, is not of key interest. In this mouse i.p. setting, a great advantage lay in conjugating avidin instead of streptavidin to the MAb. We expected the glycosylated avidin to give the unbound pretargeting molecules an intrinsic ability to rapidly progress to the liver where they would be degraded. Our data from the animal studies supported this theory. Moreover, taken together with the in vitro results, which showed that avidin-MX35-conjugate was retained on the surface of OVCAR-3 cells, we can draw some conclusions regarding the time between injections of pretargeting and effector molecules. Overall, the decrease in cell-associated effector molecules over time was moderate; this indicated that a sufficient amount of the pretargeting molecules remained on the cell surface for at least 48 hours. In addition, preliminary studies of the in vivo distribution of 211At-B-PLsuc at different times after avidin-MX35 administration showed an increase in activity in the kidneys up to 24 hours. After that, the levels were comparable to those that resulted from a single injection of 211At-B-PLsuc (data not shown). This indicated that, after 24 hours, any residual pretargeting molecules had a negligible effect. Hence, based on the in vivo and in vitro behavior of the avidin-MX35-conjugate, we concluded that 24 hours was an appropriate time period between the two injections.
Conclusions
The results from this study provide a good basis for future i.p. therapy studies of microscopic ovarian cancer in mice. We found that blood data alone were not sufficient for estimating the absorbed dose to bone marrow, but based on RBM uptake and WBC counts we concluded that at least 1.0 MBq of 211At-B-PLsuc can be safely administered 24 hours after i.p. injection of avidin-MX35 to achieve a favorable distribution of activity and avoid major toxicities. In addition, α-camera imaging clearly shows a specific uptake of 211At-B-PLsuc in avidin-MX35 pretargeted tumors, at a level that is comparable to that of conventional RIT.
Footnotes
Acknowledgments
The authors wish to thank Helena Kahu at the Department of Oncology, University of Gothenburg, Sweden, for excellent technical assistance with the animal experiments and tumor cell culturing. At the same department, we acknowledge Dr. Jörgen Elgqvist for his contributions regarding both theoretical and practical matters.
This work was supported by grants from the Swedish Cancer Society, the King Gustaf V Jubilee Clinic Research Foundation in Gothenburg, Sweden, and the Swedish Research Council. The authors declare that they have no conflict of interest.
Disclosure Statement
There are no existing financial conflicts.