
Abstract
Radioresponse is influenced by factors apart from the targeted cancer cells; in fact, endothelial cells and infiltrating immune cells within the tumor microenvironment (TME) are the two main components affecting the outcome of radiotherapy. The benefits of fractionated radiotherapy are attenuated through the upregulation of hypoxia inducible factor-1α and vascular endothelial growth factor. The therapeutic effect of antiangiogenic agents is counteracted by the mobilization of endogenous proangiogenic cells to the TME. This study highlights the importance of radiation timing within a vascular normalization window and discusses the importance of immune cells that comprise the microenvironment. A balance between favorable tumor-infiltrating immune cells, including cytotoxic T cells, natural killer cells, and dendritic cells, and the unfavorable cells, such as tumor-associated macrophages and regulatory T cells, determines the final tumor-control probability. The reciprocal complementation between combinations of radiotherapy and immunotherapy strategies through modulation of the tumor immunological microenvironment may yield promising results in the future.
Introduction
Before investigating radiosensitization, it is important to first define it and ultimately distinguish radiosensitivity from radioresponse. Radiosensitivity is defined as the susceptibility of in vitro cultured cell lines to radiation and is determined by observing the effect of radiation on colony formation. Radioresponse, however, refers to the in vivo changes in tumor size through the shrinkage or growth retardation after radiotherapy. The fact that radioresponse is closely associated with the tumor microenvironment (TME) was a significant finding in radiosensitization research. 1,2 The most commonly used radiosensitizers are chemotherapeutic agents, with the therapeutic index determined by the radiosensitization between tumor tissues and their surrounding normal tissue being within the range of a few centimeters. Radiotherapy is not deemed beneficial if the radiosensitivity of normal tissues also increases after sensitization. Microcosmically, radioresponse must take the TME condition (several μm to mm in diameter) and tumor cell radiosensitivity into account, as both factors influence the final result of radiotherapy. The two most notable TME components affecting the efficacy of radiotherapy are endothelial and immune cells. This review discusses the following: the schedule of radiotherapy during antiangiogenesis treatment, the immune-potentiating effect of targeted therapy, the importance of p53 in TME, the direct radiosensitization effect from immune cells, and the methods responsible for transforming local apoptosis into systemic immunity.
Antiangiogenesis and Radiosensitization
In 2003, Garcia-Barros et al. 3 reported that tumors in apoptosis-free endothelial vessels grew faster than those growing in apoptosis-susceptible endothelial vessels. This phenomenon was also observed in different cell lines. Moreover, tumor susceptibility to radiation treatment has been shown to be contingent on vessel conditions: when a single large dose of radiation was administered to tumors in different endothelial environments, response was observed in an apoptosis-susceptible endothelial cell environment. In contrast, tumor growth remained uninhibited in endothelial cells that were unlikely to become apoptotic. Therefore, a clinically significant relationship exists between the apoptotic potential of endothelial cells and radiotherapy outcomes. 4,5
Kolesnick and Fuks 6 argued that endothelial vessels are highly sensitive cells that secrete sphingomyelinase, the enzyme acid responsible for synthesizing ceramide. Ceramide contains a sphingosine chain connected to a fatty acid and has been linked to the induction of rapid apoptosis. 7 The high radiosensitivity of vascular endothelial cells can be attributed to ceramide enrichment. Vascular endothelial growth factor (VEGF) promotes survival of endothelial cells after radiation, and the upstream regulator of VEGF, hypoxia inducible factor-1α (HIF-1α), can be upregulated under hypoxic conditions or fractionated radiotherapy. 8 HIF-1α activation initiates an adaptive response in microvascular networks during the course of radiotherapy, which attenuates radiation-induced apoptosis in these endothelial cells. 9 Therefore, the presence of HIF-1α in the microenvironment is a key determinant for radiotherapy success. To enhance the effects of radiation, strategies to prevent vascular proliferation have been recommended, including anti-VEGF antibodies, small molecule inhibitors of the VEGF receptor (VEGFR), vascular disruptive agents that act directly on the vessels, and metronomic chemotherapy. 10 –12
Schedule of Radiotherapy When Combined with Antiangiogenic Therapy
Numerous studies have shown that decreased pressure in tumor vessels, increased blood flow and tissue-oxygen content, and normalized vascular morphology are all achieved shortly after the administration of antiangiogenic therapies. 13,14 A normal distribution of vessels exists through a balance between antiangiogenesis and proangiogenesis. However, areas of hypoxia may increase if antiangiogenesis signals predominate, 14 with temporary normalization reached when antiangiogenic drugs are used to rebalance pro- and antiangiogenic signals. Clinical evidence suggests the existence of a normalization window, 15 which is a concept where VEGF therapy can normalize the vasculature within a tumor. Researchers investigating the effects of cediranib, a VEGFR inhibitor, on glioblastoma multiforme determined a significant reduction in vascular volume, as measured by MRI, within 28 days of treatment. 16 Vascular distribution, however, returned to pretreatment status after 28 days, suggesting a normalization window of less than 1 month. Dings et al. found that 1–2 days of angiogenesis inhibitor administration followed by 3 days of radiation therapy produced the most significant inhibitory effects on tumors in vivo. The therapeutic outcome was actually less desirable when the animals received angiogenesis inhibitors for 5 days followed by radiation, 17 suggesting that administration of these inhibitors for a short duration followed by radiation is the most beneficial regimen.
The present study proposes three hypotheses: first, the period of antiangiogenesis monotherapy alone should be for a short duration prior to the initiation of radiotherapy; second, radiotherapy should, ideally, be completed before the vascular normalization window ends, which is approximately 28 days long; and third, after discontinuation of radiotherapy, an angiogenic blockade should be continued to prevent vascular regrowth and tumor recurrence. A clinical study was conducted in patients with hepatocellular carcinoma who received the angiogenesis inhibitor sunitinib at 1 week prior to radiotherapy, followed by concomitant hypofractionated radiotherapy (350 cGy per faction for 15 fractions over 3 weeks). 18 A 74% response rate and 70% 1-year overall survival were observed, which is a testament to the benefit of this approach. Continuous antiangiogenesis treatment after radiotherapy more than doubled the time to tumor progression in patients when compared with discontinuing antiangiogenesis treatment at 2 weeks after radiotherapy (10 vs. 4 months, respectively) in these patients. However, as this small study is only preliminary, a more comprehensive future trial is needed to optimize the schedule of integrated radiotherapy with antiangiogenesis treatment.
Improved TME by Targeted Therapy
There have been many successful clinical demonstrations of combined targeted therapy and radiotherapy. 19,20 Inhibition of epidermal growth factor receptor (EGFR) signaling, either by EGFR antibodies that block upstream signaling or by small-molecule drugs that block downstream signaling, may reduce tumor growth, cellular activity, and angiogenesis, while indirectly changing the surrounding microenvironment. 21 Evidence has suggested that cetuximab-mediated natural killer (NK) cell-dependent cytotoxicity determines the efficacy of cetuximab on head and neck cancer cells. 22 The combinatorial use of erlotinib and bevacizumab, or sunitinib and cetuximab, had a significant inhibitory effect on radiotherapy-induced vascular proliferation. 23,24 In addition to antiangiogenesis, immunomodulation has been closely associated with the therapeutic effects of many targeted therapy agents. 25 Because of tumor heterogeneity, targeted therapy may selectively spare resistant cancer cells eventually if used alone. In addition, the chance of developing drug resistance may decrease if the strategy of early integration radiotherapy is applied. 26
p53 and Radiosensitization
p53, one of the most important tumor suppressor genes, not only protects other genes from mutagenesis, but also activates DNA repair mechanisms and induces apoptosis when damage is not repairable. In cancer cells, normal expression of p53 is associated with relatively adequate radiosensitivity, whereas aberrant p53 expression generally inhibits a radioresponse. 27,28 The magnitude of p53 expression in tumors may therefore serve as a radioresponse predictor. 29 Local injection of recombinant adenovirus-p53 in combination with radiotherapy may effectively increase the survival of patients with nasopharyngeal carcinoma, regardless of p53 expression status in the tumor. 30 Intratumoral injection of p53 does not lead to uniform distribution of the protein in all tumor cells; therefore, changes in the TME may account for the radiosensitization effect from p53-adenovirus treatment. Adenoviral infection may upregulate the expression of interferon-inducible chemokines and recruit NK and dendritic cells to the tumor. 31 A strong immune response with extensive apoptosis was observed in follicular lymphoma patients who only received very low doses of radiotherapy, and parallel gene expression profiling showed efficient p53 pathway activation. 32 A liver tumor mouse model exhibited restoration of normal p53 as a primary response, which was associated with the upregulation of innate immunity and cellular senescence. 33 Radiation-induced DNA damage may induce apoptosis by activating the p53 pathway and triggering the production of NKG2D in cancer cells, thereby initiating attacks from NK cells. Thus, immune responses and p53-induced apoptosis can be viewed as independent occurrences, but operate in coordination. 34 A therapeutic scheme combining the p53 adenovirus and radiotherapy warrants further investigation.
Intratumoral Immunity and Radiosensitization
In addition to endothelial cells, immune cells play an important role in the microenvironment that regulates tumor growth. The TME contains various cells that release cytokines to regulate immune enhancement or suppression. 35 The immune status is closely associated with treatment outcomes and cancer prognosis. For example, tumor-infiltrating lymphocytes have been reported as good prognosis indicators of many cancers, specifically melanoma, breast, head and neck, colon, and ovarian cancer. 36,37 Pages et al. 38 found that the immune contexture, as defined by the nature, density, functional orientation, and location of immune cells, is important for predicting clinical outcome. Innate (such as dendritic cells [DC] and NK cells) or specific immune effectors (such as T cells that produce interferon-gamma) are regarded as favorable immune cells that can recognize and eliminate cancer cells. 39 Gr-1+ CD11b+ myeloid-derived suppressor cells (MDSCs), M2 macrophages, neutrophils, B lymphocytes, and CD4+ CD25+FOXP3+ regulatory T cells all promote immune suppression and are thus considered unfavorable immune cells in TME. 40 Although immune infiltrates have well-known prognostic value, only a few reports have addressed whether they affect radiation responses. Apetoh et al. 41,42 found that the treatment responses of tumor implants in immunocompetent mice were greater than implants in immunodeficient hosts. Host immunity in the TME is orchestrated by tumor evolution and therapeutic response. 43,44 Tumors evolve through immune editing, and it has been shown that sustained tumor regression in myc oncogenic mouse models can only be achieved through CD4+ T-cell activation and oncogene inactivation. 45 Immune contexture is modulated by tumors, and treatment outcome is influenced by immunity. Both regulatory T cells and tumor-associated macrophages are tightly associated with MDSCs. Tumors secrete cytokines (including CSF, GM-CSF, TGF-β, IL-10, and VEGF) and various chemokines to attract MDSCs to the TME. 46 In addition, MDSCs may be a factor involved in tumor progression and treatment failure from chemotherapy, radiotherapy, immunotherapy, and targeted therapy. 47 –49
NK cells are immune cells that have had the best characterization of radiosensitization effects. Mason et al. 50 found that the Toll-like receptor 9 ligand, which is an oligodeoxynucleotide containing unmethylated CpG motifs (characteristic of bacterial DNA), may enhance radioresponses through NK activation. Neoadjuvant CpG and Herceptin treatment followed by radiation has shown better efficacy than an initial radiation treatment and subsequent adjuvant administration. 51 The mechanisms by which NK cells enhance radiation-induced cytotoxicity and thereby render irradiated tumors more vulnerable to immune attack remain to be discerned.
Treatment-Induced Immunity
An uncontrolled tumor can eventually evade the immune system. An effective treatment would reduce tumor burden and turn an unfavorable TME into a favorable one. Therapeutic success may break the vicious cycle of immunosuppressive factors, including cytokines (e.g., IL-10, TGF-β), regulatory T cells, and MDSCs throughout the TME. 52 Neoadjuvant chemotherapy has been shown to increase cytotoxic T-cell infiltration in breast cancers and cause tumor-specific immune responses. 53 Similarly, the number of specific cytotoxic T cells to survivin increases after radiotherapy in colorectal cancers. 54 Apetoh et al. 55 showed that the interaction between high mobility group box 1 protein (HMGB1), which is released from dying cells, with Toll-like receptor 4 on DCs is necessary for the induction of a specific T-cell response.
The continuous improvement of the microenvironment is critical for treatment success. Marcophage CSF-1 is a chemokine that is crucial for stem cell differentiation into suppressor macrophages and endothelial cells in the TME. 56 Marcophage CSF-1 receptors (FMS), KIT, FLT3, and PDGFRα/β are receptors inhibited by sunitinib and imatinib. 57 Ozao-Choy et al. 58 showed that sunitinib inhibits c-KIT and prevents MDSCs from being attracted to the TME, which reduces the number of regulatory T cells and increases the number of beneficial IFN-γ and NK cells in tumor-infiltrated lymphocytes. The amino-biphosphonate, zolendronic acid (ZA), disrupts a vicious cycle in the bone microenvironment by reducing the bone absorption and retarding tumor growth. 59 The combination of ZA and sunitinib exhibits a promising synergy in antiangiogenesis and bone metastasis reduction. 60 A clinically relevant dose of ZA inhibits tumor-associated macrophage and angiogenesis in TME. 61,62 Our hypothesis, model, and methods of modulating TME in order to improve the radioresponse by sunitinib, low-dose chemotherapy, and metronomic ZA are shown in Figure 1.
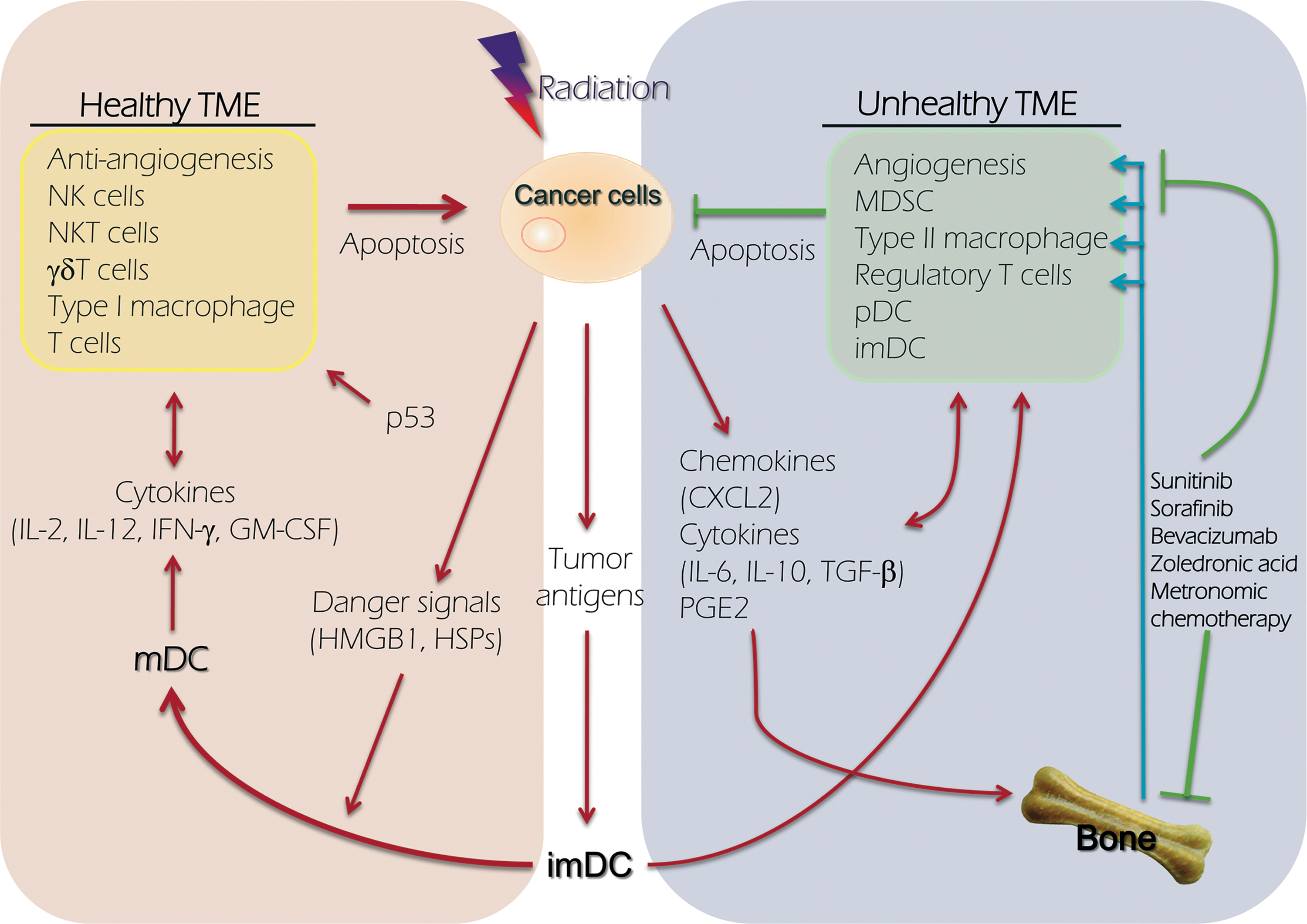
Model depicting how TME regulates cancer cell death after radiotherapy. The left part of figure represents the composition of healthy TME, which may promote the death of cancer after radiation, and the right side represents the unhealthy TME and plays an opposite function. TME, tumor microenvironment; mDC, matured dendritic cell; imDC, immaturated dendritic cell; pDC, plasmacytoid dendritic cell; MDSC, myeloid-derived suppressor cells; HSP, heat shock protein; HMGB1, high-mobility group protein B1; PGE2, prostaglandin E2.
Turning Local Apoptosis into Systemic Immunity
The abscopal effect, or eradication of a primary tumor, may either promote or inhibit the growth of metastatic tumors during radiotherapy, 63 which is dependent on the immune response at the specific site. Danger signals such as HMGB1, generated after radiotherapy, may activate immature DCs at tumor sites. T cells, however, are not efficiently primed in the draining lymph nodes if radiotherapy or intratumoral DC therapy is used alone. 64 Radiation was shown to enhance tumor antigen uptake and presentation by DCs and promoted the migration of DCs out of the tumor mass and into lymph nodes. This therapeutic was found to be further enhanced by heat shock protein (Hsp) co-administration (unpublished data). An abscopal effect has been noted when a combination of radiation and DC-based immunotherapy was used in patients with hepatoma. 65 A strategy of the modifications aimed at turning local apoptosis into systemic immunity is illustrated in Figure 2.
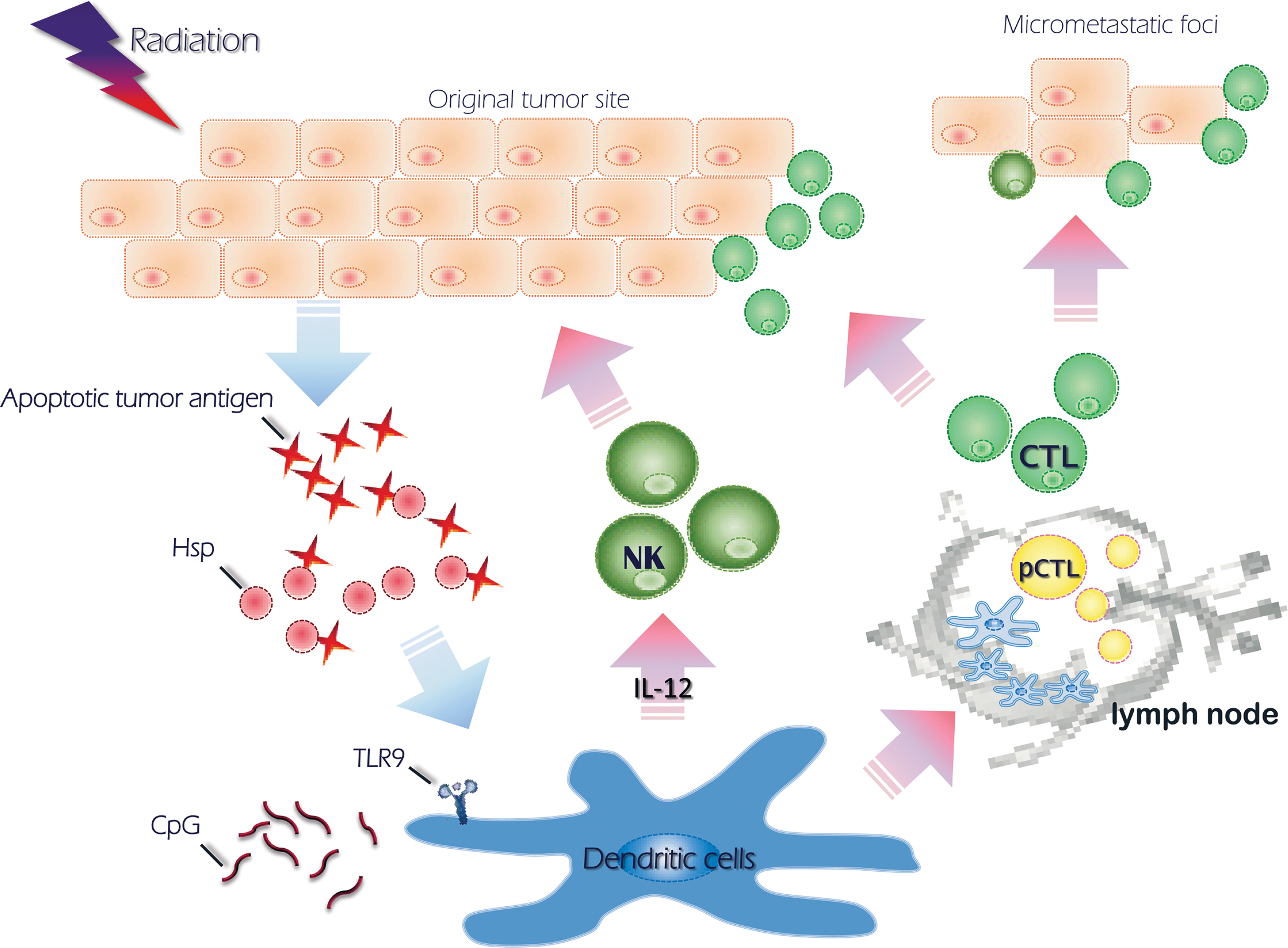
Abscopal effect from local irradiation. The diagram shows the induction of systemic cytotoxic T cells (CTL) from local radiotherapy.
Conclusions
The microenvironment is a critical aspect for tumor cell survival, death, and immunogenicity after radiotherapy. Mounting evidence supports the hypothesis that the milieu of the TME will deteriorate with disease progression unless a successful treatment can alter this cycle. Strategies have been developed for improving radioresponse through TME modulation. An understanding of the crosstalk between irradiated tumor cells and immune elements may elucidate mechanisms for turning local apoptosis into systemic immunity.
Footnotes
Disclosure Statement
There are no financial disclosures from any authors.