
Abstract
This study investigated the feasibility of targeting the free, unbound forms of prostate-specific antigen (fPSA) for in vivo imaging of prostate adenocarcinomas (PCa), as PSA is produced and secreted at abundance during every clinical stage and grade of PCa, including castration-resistant disease. We injected 125I-labeled monoclonal antibody PSA30 (specific for an epitope uniquely accessible on fPSA alone) intravenously in male nude mice carrying subcutaneous xenografts of LNCaP tumors (n=36). Mice were sacrificed over a time course from 4 hours to 13 days after injecting 125I-labeled PSA30. Tissue uptake of 125I-PSA30 at 48 and 168 hours after intravenous injection was compared with two clinically used positron emission tomography radiopharmaceuticals, 18F-fluoro-deoxy-glucose (18F-FDG) or 18F-choline, in cryosections using Digital AutoRadiography (DAR) and also compared with immunohistochemical staining of PSA and histopathology. On DAR, the areas with high 125I-PSA30 uptake corresponded mainly to morphologically intact and PSA-producing LNCaP cells, but did not associate with the areas of high uptake of either 18F-FDG or 18F-choline. Biodistribution of 125I-PSA30 measured in dissected organs ex vivo during 4 to 312 hours after intravenous injection demonstrated maximum selective tumor uptake 24–48 hours after antibody injection. Our data showed selective uptake in vivo of a monoclonal antibody highly specific for fPSA in LNCaP cells. Hence, in vivo imaging of fPSA may be feasible with putative usefulness in disseminated PCa.
Introduction
Imaging modalities used for the detection and monitoring of extraprostatic or locally advanced growth of prostate adenocarcinomas (PCas) are constantly developing and improving. 1,2 Apart from morphological imaging, such as ultrasonography, computed tomography, and magnetic resonance imaging (MRI), molecular imaging like positron emission tomography (PET) imaging of metabolic probes, such as 18F-fluoro-deoxy-glucose (18F-FDG) and 18F- or 11C-labeled choline, has shown to improve detection of both locally advanced and metastatic disease. 18FDG measures glucose metabolism, which is upregulated in many cancers. However, most primary PCa tumors exhibit low, if any, 18FDG uptake. 3 –6 On the other hand, choline kinase is frequently overexpressed in PCa 7,8 and as 18F- or 11C-labeled choline derivatives are trapped inside the cell by choline kinase, information is provided about the rate of lipid synthesis. 9 –13 However, there is also uptake of both 18F- and 11C-labeled choline in benign disease conditions as well. 14
Prostate-specific antigen (PSA) is the gene product of KLK3, which is one of 15 kallikrein-related peptidase genes (KLK1-KLK15) on chromosome 19q13.4. 15,16 PSA released into the blood is noncatalytic, in part due to inactivation subsequent to the formation of stable covalent complexes with major extracellular protease inhibitors, particularly α-1-antichymotrypsin (SERPINA3 or ACT), although reactions with α-2-macroglobulin (A2M) are faster, at least in vitro. 17,18 A smaller percentage (5%–40%) of the noncatalytic PSA in the blood occurs as free, unbound forms (free PSA [fPSA]) that are unable to form complexes with, for example, ACT or A2M despite ≈104-fold excess of these and other protease inhibitors in the blood. 17,19 While fPSA (28.4 kDa) is eliminated by a half-life of 12–18 hours via glomerular filtration by the kidneys, by contrast, the ≈90 kDa PSA-ACT complex (cPSA) is too large to allow for renal clearance and is eliminated very slowly, possibly via uptake by hepatocytes. 20 –24
Prior attempts to target PSA for in vivo imaging of PCa metastases investigated human subjects with advanced disseminated stages of the disease 25 ; only one study was based on mice xenografts. 26 Imaging of PSA was mostly successful in these studies; however, the image quality was poor due to high liver uptake and high nonspecific background activity. 27 Notably, the design of these studies was not based on the subsequently reported investigations showing that PSA in the extracellular fluids occurs in many different molecular forms with distinctly different rates and mechanisms of clearance. 17,20,28,29 Also, the antibodies used in prior studies to detect PSA in vivo were polyclonal; hence, they could cross-react with other antigens and did not discriminate fPSA from cPSA. This feat was not possible until the early 1990s when it was first reported on the discovery of fPSA and the development of monoclonal antibodies specific to antigenic epitopes uniquely accessible on fPSA alone, but unable to detect PSA linked to protease inhibitors, such as ACT. 17,19,30
Therefore, as no prior study explored the feasibility of using fPSA as a target for in vivo imaging, we now investigated whether a monoclonal antibody (mAb) specific for fPSA [PSA30] alone may prove to be a useful candidate to image advanced and metastatic PCa in vivo. Digital AutoRadiography (DAR) was used to observe specific PSA30 uptake in viable PSA-secreting PCa cells, and a biodistribution study was done to reveal the biokinetics in healthy and tumor-bearing animals. As a comparison to clinically used metabolic tracers, we co-injected PET radiopharmaceuticals, 18F-FDG or 18F-choline, with 125I-labeled mAb PSA30 in animals chosen for DAR analysis. From these tests, we were able to distinguish different patterns of uptake in tumor sections by isotope separation using DAR images corresponding to histology and immunohistochemistry (IHC) of adjacent sections.
Materials and Methods
The PSA30 mAb
To assess whether it may be feasible to use human fPSA as a target for the imaging of PCa cells in vivo, we used the PSA30 anti-fPSA mouse mAb, which has an IgG1 isotype as previously described. 31 Comprehensive epitope mapping studies have demonstrated that PSA30 has very similar binding characteristics to some of the originally developed mouse mAbs (e.g., 5A10) specific for fPSA alone, 17,19,30,32 and PSA30 recognizes an epitope covered by ACT that is accessible only on fPSA. 32,33
Radiolabeling
The PSA30 antibody was labeled with 125I (PerkinElmer), using the Iodogen method. Briefly, a coated test tube with 150 μg 1,3,4,6-tetrachloro-3α,6α-diphenyl glycoluril was used for labeling of 200 μg PSA30. After the mixture had been incubated for 15 minutes at room temperature, low-molecular-weight components were removed by gel filtration (PD-10 column; GE Healthcare). The radiochemical purity was 95% after gel filtration.
The 2-18FDG and [18F]Fluoromethylcholine were produced by an in-house cyclotron. The radiochemical purity for both of the radiopharmaceuticals was >99%.
mAb-based immunoradiometric assays (IRMAs) for 125I-PSA30 binding quality were conducted in triplicate as a four-step sandwich assay with wash steps between incubations (washing buffer: 10 mM Tris-HCL [pH 8.0], 0.15 M NaCl, and 0.05% Tween 20). The assay was constructed and optimized according to established recommendations. 34 Break-apart microtiter plates were coated with H117 (0.2 μg/well), a mAb recognizing free or total PSA and human kallikrein 2 (hK2) with the same affinity 30 ; diluted in coating buffer (75 mM sodium carbonate [pH 9.6]); and incubated overnight at 4°C. The wells were then incubated with 0.2 μg/well quenching buffer (3% fish gelatin in washing buffer) for 2 hours at room temperature. Next, the wells were coated with 200 μL plasma (female) containing 3 ng/μL fPSA and incubated for 2 hours at room temperature. 125I-labeled and unlabeled PSA30 were then mixed together in assay buffer (50 mM Tris-HCl [pH 7.5], 0.1 M NaCl, 5 mM EDTA, 0.25% BSA, and 0.05% Tween 20) at descending concentrations and added to the wells (total volume: 50 μL/well). The percentage of labeled antibody per well was as follows: 100%, 92%, 84%, 68%, 50%, 30%, and 0%. The plates were incubated for 2 hours at room temperature, washed, and measured in a NaI(Tl)–well counter (1282 Compugamma CS; LKB Wallac). A difference in detection capacity of <25% in relation to theoretical deviance was accepted for further application. The estimations of detection quality postlabeling showed that 125I-labeled PSA30 maintained 87% of the affinity/binding capacity of the unlabeled PSA30 antibody.
Cell culture and animal models
Male athymic nude mice NMRI-NU (nu/nu), aged between 6 and 8 weeks, were purchased from Taconic Europe. Only animals showing progressive weight gain and weighing more than 18 g before inoculation were used. The mice were provided sterilized food and water ad libitum and housed in individually ventilated cages under sterile conditions. All animal experiments were conducted in accordance with protocols prepared and approved according to the guidelines set by the Malmö-Lund Ethical Committee for the use and care of laboratory animals.
LNCaP cells (ATCC) were grown as a monolayer in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin streptomycin. The cells were maintained at 37°C in an atmosphere of air with 5% CO2. LNCaP cells, harvested in 0.02% trypsin/phosphate buffered saline (PBS), were resuspended in media and injected subcutaneously into the right flank with 200 μL of cell suspension (∼2×106 tumor cells) containing an equal mix of 100 μL of Matrigel (BD Biosciences) and cells (100 μL) on ice. Tumor formation was monitored visually and by palpation.
Tumor section imaging
LNCaP-tumor-bearing mice (n=36) were administered 125I-labeled PSA30 formulations (∼10 MBq [27 μCi], 15 μg of PSA30 in 50 μL sterile saline) and 18F-choline (n=2) or 18F-FDG (n=5) via injection into the tail vein. Mice were allowed free movement after all injections. Access of metabolic probes was dependent on surplus from patient studies. 18F-choline was administered 48 hours and 18F-FDG was administered 24, 72, 168, and 312 hours postinjection of mAb. Animals were euthanized one hour postinjection of metabolic probes and tumors were immediately removed, secured in Cryomount (HistoLab Products AB), quickly frozen in liquid nitrogen, and cut into 100-μm sections for DAR or 20-μm sections for histopathology and IHC analysis. A silicon strip detector based system (Biomolex 700 Imager; Bimolex AS) was used to image the distribution of radioactivity within the thicker sections. Differences in both emission spectra and rate of decay were used to produce separate images of each radionuclide in animals injected with more than one radionuclide, in this case, 125I and 18F.
IHC and histopathology
To study PSA expression, 20-μm tumor cryosections (frozen and secured as described previously) were examined using IHC. The immunoreactivity against PSA was visualized by use of the DAKO EnVision Flex/HRP system kit (Dako Corporation). Adjacent tumor sections were also stained with hematoxylin (nuclei stain) and eosin (cytoplasmic stain) (H&E) and the general morphology was analyzed under a standard transillumination microscope. With H&E staining, viable regions of the tumor sections and necrotic areas were stained. As a positive control, LNCaP tumor sections were incubated with PSA mAb 2E917 at a dilution of 1:1000 and visualized as described previously. As a negative control, tumor section from a mouse that received an intravenous injection of PSA30 was visualized without incubation of a secondary antibody, but including all other steps of IHC. The stained sections were scanned using a Carl Zeiss MIRAX Scan microscope scanner and viewed with the MIRAX Viewer software (Carl Zeiss Imaging Solutions GmbH).
Biodistribution study
Biodistribution studies were conducted in both nontumor-bearing mice (n=14) and in LNCaP-tumor-bearing mice (n=34). All mice were randomized before the study and administered 125I-PSA30 (∼10 MBq [27 μCi], 15 μg of PSA30 in 50 μL sterile saline for injection) via injection into the tail vein (0 hour). LNCaP-tumor-bearing mice were euthanized after 4, 24, 72, 168, and 312 hours, and nontumor-bearing animals at 6, 24, 96, 168, and 312 hours after injection. Eleven organs (including tumor) were removed, and collected in preweighed tubes and weighed again, and the activity was measured in a NaI(Tl)–well counter (1282 Compugamma CS; LKB Wallac). Our results are presented as decay-corrected percentage of injected activity per gram of tissue (%IA/g), organ-to-blood (O/Bl) and tumor-to-organ (T/O) ratios, and as mean values of parameters±standard deviation.
Results
DAR, IHC, and histopathology
PSA30 was labeled with 125I and studied for in vivo targeting of fPSA in LNCaP xenograft models. Tumor sections from the LNCaP-xenografted mice were imaged with DAR. The DAR images shown in Figure 1 are taken from the same LNCaP xenograft tumor section. These pictures show the distribution of 125I-PSA30 at 48 hours postinjection (Fig. 1A) and 18F-choline at 1 hour postinjection (Fig. 1B) accompanied by adjacent sections of H&E and IHC staining for PSA. Similarly, Figure 2 shows the DAR images of 125I-PSA30 (168 hours postinjection) and 18F-FDG (1 hour postinjection) activities, respectively, in another LNCaP-based xenograft tumor section accompanied by H&E in an adjacent section of the tumor. These DAR images show uniform distribution of 125I-PSA30 in tumor sections containing densely packed viable cells—viable in respect to the maintenance of PSA production as confirmed by IHC and preserved morphological features in H&E staining. In particular, high activity of 125I-PSA30 was manifested in proximity to blood vessels, capillaries, and areas with viable PSA-secreting tumor cells (Fig. 1H). In areas of microscopically well-preserved cells, there was little, if any, association between high activity of 125I-PSA30 compared with the uptake of 18F-choline or 18F-FDG (Figs. 1 and 2). Notably, as the mice were allowed free movement after the 18F-FDG injection, as expected, the DAR images showed a high degree of 18F-FDG uptake in muscle (Fig. 2, red square) that was left on the tumor after it was removed from the mouse; this uptake is not seen on the 125I-PSA30 DAR picture of the same tumor. Except for areas of necrosis, there was close similarity between the distribution of PSA staining by IHC and high activity of 125I-PSA30 uptake on DAR, thus confirming that the radioactivity found in tumor sections by DAR was strongly associated with evidence of PSA staining by IHC and not a consequence of the presence of free iodine or other metabolites.
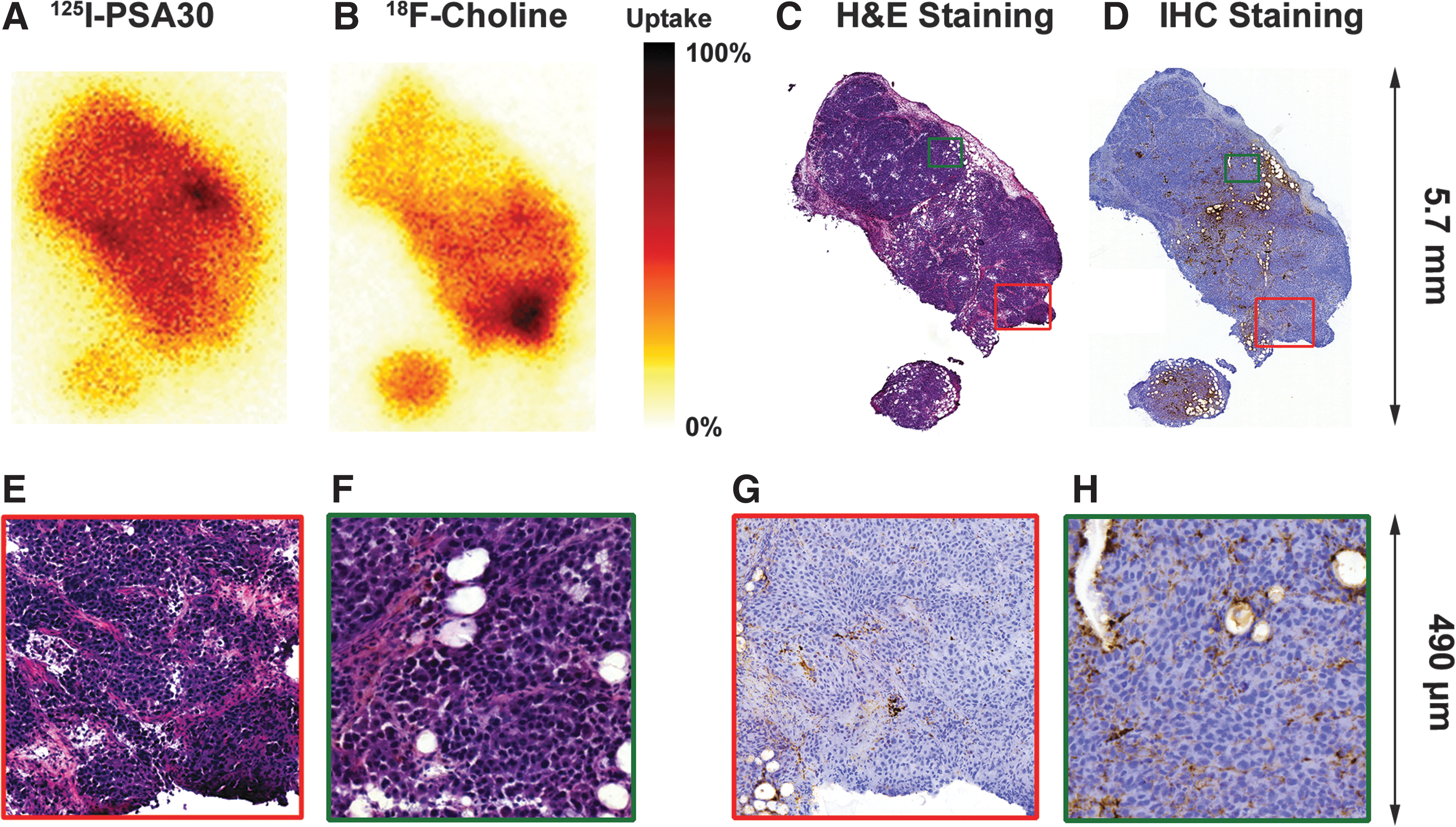
DAR: Individually normalized uptake of 125I-PSA30

DAR: Individually normalized uptake of 125I-PSA30 and 18F-FDG (168 hours postinjection of 125I-PSA30 plus 1 hour postinjection of 18F-FDG) in the same tumor section separated by isotope. Histological analysis was through H&E-stained adjacent sections. There is no direct association between areas of high PSA30 mAb uptake and high FDG uptake. However, as expected since the mouse was not kept sedated after the 18F-FDG injection, there is a high degree of FDG uptake in tissue/muscle (red square) left after the tumor was removed from the mouse; this uptake is not seen on the 125I-PSA30 DAR picture. 18F-FDG, 18F-fluoro-deoxy-glucose.
Biodistribution in nontumor-bearing mice
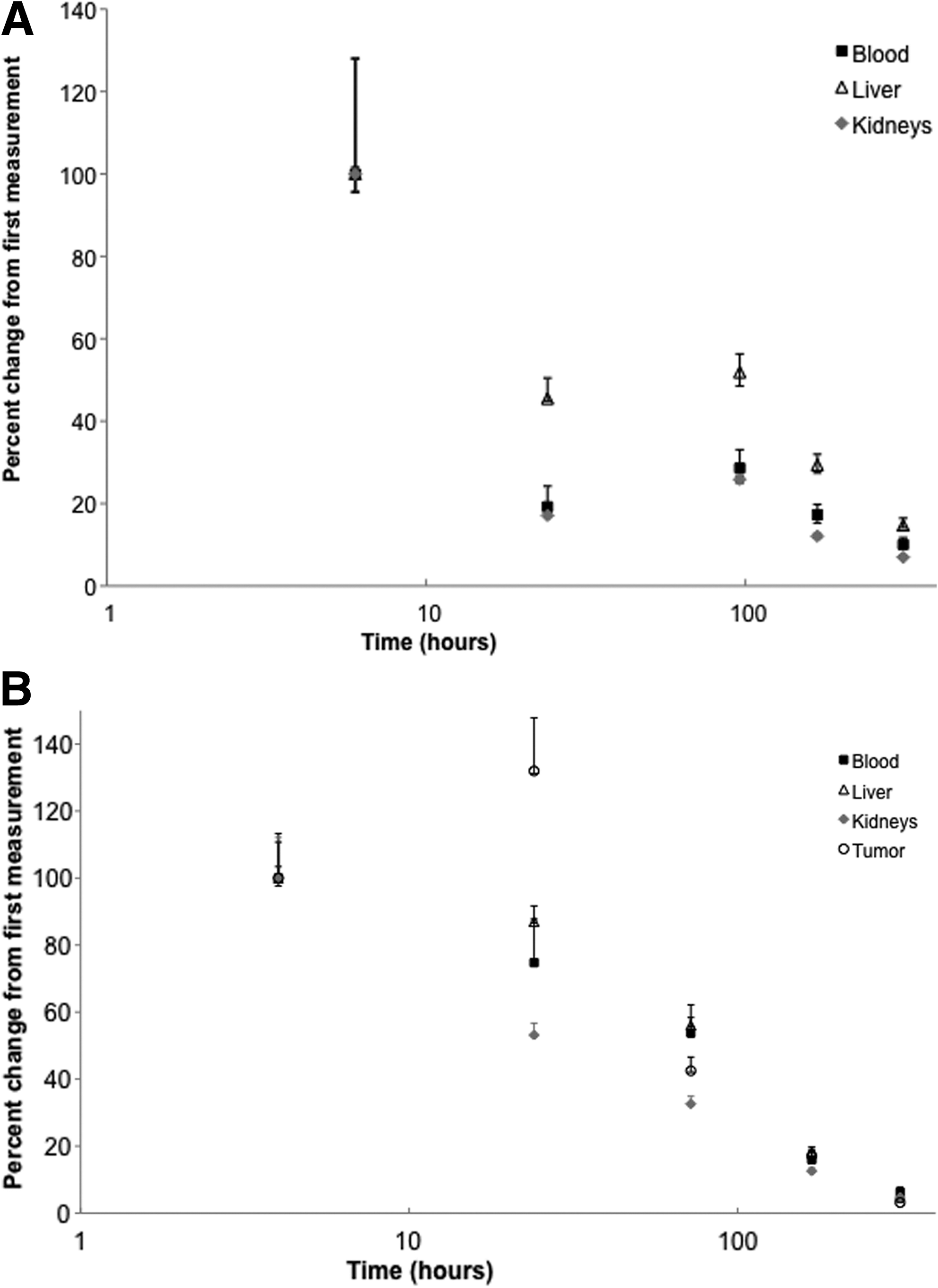
A decrease of activity after injection was noted in all organs (except the thyroid), and blood from the first measurement after intravenous injection of the 125I-PSA30 formulations to the last (312 hours) (see Table 1A). As shown in Figure 3A, the decrease in %IA/g during the first 24 hours ranged from 55% to 83% in liver, kidney, and blood (Fig. 3A). The highest %IA/g was found in organs with large residual blood content. 35 The O/Bl ratios in highly vascularized organs (see Table 2) remained largely unchanged over time, indicating that no unspecific 125I-PSA30 binding occurred, which is in accordance with the fact that PSA is not expressed in nonprimate species, such as mice and rodents. 36 However, thyroid levels increased from 6 to 168 hours postinjection.
Mean±standard deviation.
Thyroid data presented as % IA per whole organ, not per gram.
PSA, prostate-specific antigen; % IA/g, percentage of injected activity per gram of tissue.
Biodistribution in LNCaP-tumor-bearing mice
Similar to our findings in nontumor-bearing mice, highly vascularized organs initially showed high activity levels that rapidly decreased from the first measurement after intravenous injection of 125I-PSA30 (see Table 1B; Fig. 3B). Similarly, the O/Bl ratios remained largely unchanged over time in these organs (see Table 2). LNCaP tumors had higher uptake compared with other investigated organs at most time points and peaked (4.32 %IA/g) at 24 hours after intravenous injection of 125I-PSA30 formulations. By contrast to all other organs showing a decrease of activity, LNCaP tumors showed a marked increase of activity (by 32%) during the first 24 hours after injection (Fig. 3B). Tumor-specific accumulation of 125I-PSA30 was further demonstrated by a markedly divergent O/Bl ratio (see Table 2). In comparison to nontumor-bearing mice, thyroid accumulation was greatly augmented. 125I-PSA30 mAb uptake in LNCaP tumors peaks at 24 hours postinjection (Fig. 3B), with a subsequent sharp decrease in tumor uptake noted by 72 hours postinjection. Importantly, at this same time point, there is a sharp increase in thyroid uptake. This inverse correlation is a likely indicator that a dehalogenation effect has occurred.
Mean±standard deviation.
Thyroid data presented as % IA per whole organ, not per gram.
Discussion
The use of PSA as an in vivo imaging target was previously investigated 15–20 years ago, but abandoned as an imaging target due to high activity in blood and large uptake in blood-bearing organs. 25,26,37 However, advances in the field of antibody manufacturing combined with better understanding of the structure–function relationship of PSA and its interaction with protease inhibitors prompted us to hypothesize that the targeting of fPSA, rather than complexed or total PSA, could be present at high abundance in close proximity to its local site of production. This could provide another theoretical advantage as fPSA has been shown to clear rapidly (t 1/2 of 12–18 hours) from the extracellular compartments by glomerular filtration in the kidneys, 20,23,24 as opposed to the PSA-ACT complex that is too large (≈90 kDa) to allow for renal clearance. Hence, the elimination rate of PSA-ACT (and also total PSA) is slow with possible limited capacity. 23
Contingent of that, in vivo targeting of fPSA would be feasible and generate high and specific uptake useful for molecular imaging with PET or single-photon emission computed tomography (SPECT). The detection of fPSA would be anticipated to represent presence of both benign as well as malignant prostatic tissue, including metastatic lesions of PCa to both bone and soft tissue. The method could also facilitate the generation of images of extracapsular growth and residual prostatic tissue following any treatment given with a curative intent. The only clinical limitation we can foresee by targeting fPSA would occur in patients who have severe impairment of renal function and thus a substantial increase in the proportion of fPSA in the blood, 28 which could result in increased background signal.
To the best of our knowledge, the specific targeting of fPSA for in vivo imaging has not previously been reported. The targeting of fPSA may also be unique due to that it represents a highly abundant secretory protein, 38 as opposed to current strategies to target extracellular domains of integral plasma membrane proteins, such as the prostate-specific membrane antigen. 39 The diagnostic information contributed by a fPSA targeting strategy is likely to be influenced by mutations, other genetic rearrangements, or therapies affecting the functionality of the androgen receptor (AR) as production and secretion of fPSA is regulated by androgens and AR function. 15,40,41 In the present study, the specific targeting of fPSA was investigated by using PSA30, an IgG1 that is specific for an epitope region uniquely available on fPSA alone. 30,32 We successfully employed labeling with 125I and evaluated targeting by DAR and biodistribution studies in both LNCaP-tumor-bearing mice and nontumor-bearing animals. The uptake of 125I-PSA30 on DAR was compared with the uptake of co-injected 18F-labeled metabolic probes (FDG and choline).
Our results show that 125I-PSA30 can effectively target fPSA in LNCaP-based xenograft mice. DAR revealed a pattern of uptake associated to PSA expression. However, we also noted bright hotspots in necrotic areas where the 125I-PSA30 uptake in these necrotic areas did not correspond to PSA expression, as revealed on IHC.
We also evaluated 125I-PSA30 mAb uptake in relation to 18F-FDG or 18F-choline in the same animal and found that FDG uptake, a measure of cellular metabolic activity (Fig. 2), and choline uptake, a measure of increased cellular proliferation (Fig. 1B), did not correlate with abundant PSA production. Inflammation and necrosis were noted in the areas where 125I-PSA30 mAb and metabolic probes did correspond. Thus, this association can be attributed to two factors. First, the accumulation of the mAb [PSA30] in necrotic areas of solid tumors is generally accepted to be a result of the enhanced permeability and retention effect 42,43 ; second, it is also well documented that the major downfalls of metabolic probes, for example, 18F -choline and 18F-FDG, are their unspecificity toward malignancies but more tendency to accumulate in areas of inflammation. 44 –47 These findings demonstrate that PSA30 has a potential to serve as an independent marker for PCa imaging that might provide information above and beyond the scope of metabolic and proliferative status. This might be important because all studies indicate the heterogeneity of PCa and hence the possible need for more than one single marker. The clear distinction of uptake seen between PSA30, choline, and FDG via DAR indicates that antibodies targeting fPSA may be helpful to differentiate between tumor cells and nonspecific metabolism or proliferation that holds back current metabolic probes.
Our DAR data were supplemented with biodistribution studies (see Tables 1A, B). Based on these data, we demonstrate that the PSA30 mAb uptake in excised tumors peaked at 24 hours postintravenous injection, and is retained in tumor as compared with normal tissues (see Table 1B). The relatively low T/O ratios (see Table 3) can be attributed to factors, such as a binding site barrier, seen when a low antibody dose is saturated by the fPSA antigens in the perivascular space thus preventing deeper penetration into the solid tumor 43,48 ; insufficient vascular permeability inside of the tumor; or deiodination of the antibody (as suggested by the high iodine accumulation in the thyroid). Two ways to improve the T/O ratios would be to increase the antibody dose and test different radiolabels. Despite this drawback, we found an accumulation of 125I-PSA30 activity in tumor tissue.
Notable is the observed rapid decrease of activity from the liver and kidneys between 4 hours and 13 days (both −95%), illustrating the antibody is not retained for long periods of time in the organs. The size of both IgG1 and IgG1 bound to PSA prevents it from renal filtration. Thus, the activity observed in the kidney most likely reflects free iodine that has re-entered the circulation after the antibody has been digested in the liver or due to free iodine caused by dehalogenation.
Conclusions
Our results suggest that it is feasible to image the free, unbound forms of PSA using a radiolabeled mAb specific for fPSA, and thus the development of a means to evaluate fPSA in vivo as an imaging target in PCa should be continued. To achieve higher T/O and T/Bl ratios we now aim to increase the antibody dose administered, test alternative labeling techniques and radionuclides, as well as fragmented antibodies.
Footnotes
Acknowledgments
The authors thank Drs. NagaVaraKishore Pillarsetty (Memorial Sloan-Kettering Cancer Center, New York, NY) and Kim Pettersson (Turku University, Finland) for informative discussions. We are also grateful to Mikael Håkansson of the Institute of Mathematical Statistics, Lund University, for carrying out the statistical evaluation on the IRMA results. The study was supported by grants from Swedish Cancer Society (Cancerfonden) [AB: 11 0263; HL: 3455; S-ES: 1105 21], Swedish Research Council (Vetenskapsrådet, Medicine) [AB: A0281801; HL: 20095], Gunnar Nilsson Cancer Foundation, VINNOVA/Eurostar, The Tegger Foundation, Berta Kamprad Foundation, Lund University Medical Faculty ALF grants, the National Cancer Institute [R33 CA 127768-02 and P50-CA92629]; the Sidney Kimmel Center for Prostate and Urologic Cancers; David H. Koch through the Prostate Cancer Foundation; and Fundación Federico SA. The study sponsors had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication.
Disclosure Statement
Dr. H. Lilja holds patents to fPSA, intact PSA, and hK2 measurements in blood in vitro. Neither Dr. O. Nilsson nor Fujirebio Diagnostics has any commercial interest or has provided any financial support for the submitted work.