
Abstract
The C3-monoamine on the carbohydrate moiety (daunosamine -NH2-3′) of epirubicin was reacted under anhydrous conditions with succinimidyl 4,4-azipentanoate to create a covalent UV-photoactivated epirubicin-(C3-amide) intermediate with primary amine-reactive properties. A synthetic covalent bond between the UV-photoactivated epirubicin-(C3-amide) intermediate and the ɛ-amine of lysine residues within the amino acid sequence of anti-HER2/neu monoclonal immunoglobulin was subsequently created by exposure to UV light (354 nm) for 15 minutes. Size-separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis combined with immunodetection analysis and chemiluminescent autoradiographic imaging revealed a lack of IgG-IgG polymerization or degradative protein fragmentation of the covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic. Retained binding-avidity of epirubicin-(C3-amide)-[anti-HER2/neu] was validated by cell-ELISA utilizing monolayer populations of chemotherapeutic-resistant mammary adenocarcinoma SKBr-3 which highly overexpress membrane-associated HER2/neu complexes. Between epirubicin-equivalent concentrations of 10−10 to 10−6 M the covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic consistently evoked levels of cytotoxic anti-neoplastic potency that were highly analogous to chemotherapeutic-equivalent concentrations of epirubicin. Cytotoxic anti-neoplastic potency of epirubicin-(C3-amide)-[anti-HER2/neu] against chemotherapeutic-resistant mammary adenocarcinoma SKBr-3 challenged with epirubicin-(C3-amide)-[anti-HER2/neu] at an epirubicin-equivalent concentration of 10−6 M was 88.5% (e.g., 11.5% residual survival). Between final epirubicin-equivalent concentrations of 10−8 and 10−7 M there was a marked threshold increase in the mean cytotoxic anti-neoplastic activity for epirubicin-(C3-amide)-[anti-HER2/neu] from 9.9% to 66.9% (90.2% to 33.1% residual survival).
Introduction
Anthracyclines have traditionally been the class of chemotherapeutic most extensively utilized to synthesize covalent immunochemotherapeutics with properties of selective “targeted” delivery. 1 –26 Monoclonal immunoglobulin fractions are frequently utilized for specific recognition and physical binding to antigens or receptor complexes uniquely overexpressed on the exterior surface membrane of neoplastic cell populations. 7,26 –30 Doxorubicin 28,29,31 –33 has been the most common anthracycline to date utilized to synthesize covalent immunochemotherapeutics in addition to the relatively limited utilization of daunorubicin 34 –36 and epirubicin. 7,26,37,38 The combination of profound cytotoxic anti-neoplastic potency in addition to their chemical composition which provides multiple opportunities to create covalent bond structures collectively contribute to the popular use of anthracyclines for the synthesis of immunochemotherapeutics and receptor ligand-chemotherapeutics that utilize molecular platforms to facilitate selective “targeted” chemotherapeutic delivery. Covalent anthracycline immunochemotherapeutics have been designed to selectively “target” chemotherapeutic delivery, and evoke potent ex vivo cytotoxic anti-neoplastic potency against several neoplastic cell types including mammary adenocarcinoma (anti-HER2/neu, anti-epidermal growth factor receptor [EGFR]), 7,26 colon adenocarcinoma (anti-CEA) 36 ; multiple myeloma (CD38+, MC/CAR), 28 B-lymphoma, 32 melanoma, 27,29,35 gastric carcinoma, 39 colon carcinoma, 37 pulmonary carcinoma, 40 and other neoplastic cell types (CEA). 34,35 In direct accord with their level of in vitro efficacy, similar covalent anthracycline immunochemotherapeutics reduce in vivo tumor burden and prolong survival against human xenografts of gastric carcinoma, 39 breast cancer, 33 CD38 positive MC/CAR multiple myeloma, 28 B-lymphoma, 32 T-cell lymphoma, 41 colon carcinoma, 33,42 –44 ovarian carcinoma, 42 pulmonary carcinoma, 33 metastatic melanoma, 27,29 hepatocellular carcinoma, 31 and intracerebral small-cell lung carcinoma. 45 –47 In part these findings correlate with the recognized additive and synergistic levels of cytotoxic anti-neoplastic potency of anti-HER2/neu (inhibited HER2/neu function) in concert with conventional chemotherapeutics such as cyclophosphamide, 48,49 docetaxel, 48 doxorubicin, 48,49 etoposide, 48 methotrexate, 48 paclitaxel, 48,49 or vinblastine. 48 Similar to anti-HER2/neu, 48 –53 other trophic receptor inhibitors including anti-EGFR 54 –56 and anti-VEGFR 57 –59 also create additive and synergistic levels of cytotoxic anti-neoplastic potency when applied in combination with conventional chemotherapeutic agents.
Despite the development and use of different methods for the production of covalent anthracycline immunochemotherapeutics few reports have described the design of relatively rapid synthesis schemes that also employ gentle reaction conditions that afford a lower risk of degradation or polymerization of biological protein fractions and do not have a requirement for pre-thiolation at the ɛ-amine of lysine amino acid residues. To address this void in the literature, a methodology has been developed for the synthesis of covalent epirubicin immunochemotherapeutics utilizing a UV-photoactivated covalent epirubicin-(C3-amide) intermediate that possess primary amine-reactive properties.
Materials and Methods
Synthesis of epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic
Phase-I synthesis scheme for UV-photoactivated epirubicin-(C3-amide) intermediates
The C3 monoamine on the carbohydrate moiety of epirubicin (3.02×10−7 mg, 1.75×10−4 mmoles) was reacted at a 5:1 molar-ratio with the amine-reactive N-hydroxysuccinimide ester “leaving” complex of succinimidyl 4,4-azipentanoate (1.55×10−7 mg, 3.5×10−5 mmoles) in the presence of triethylamine (50 mM final concentration) utilizing dimethylsulfoxide as an anhydrous organic solvent system (Fig. 1). The reaction mixture formulated from stock solutions of epirubicin and succinimidyl 4,4-azipentanoate was continually stirred gently at 25°C over a 4-hour incubation period in the dark and protected from exposure to light. The relatively long incubation period of 4 hours was utilized to maximize degradation of the ester group of any residual succinimidyl 4,4-azipentanoate or succinimidyl 2-[(4,4′-azipentanamido)ethyl]-1,3′-dithioproprionate that may not have reacted in the first 30 to 60 minutes with the C3 monoamine group of epirubicin.
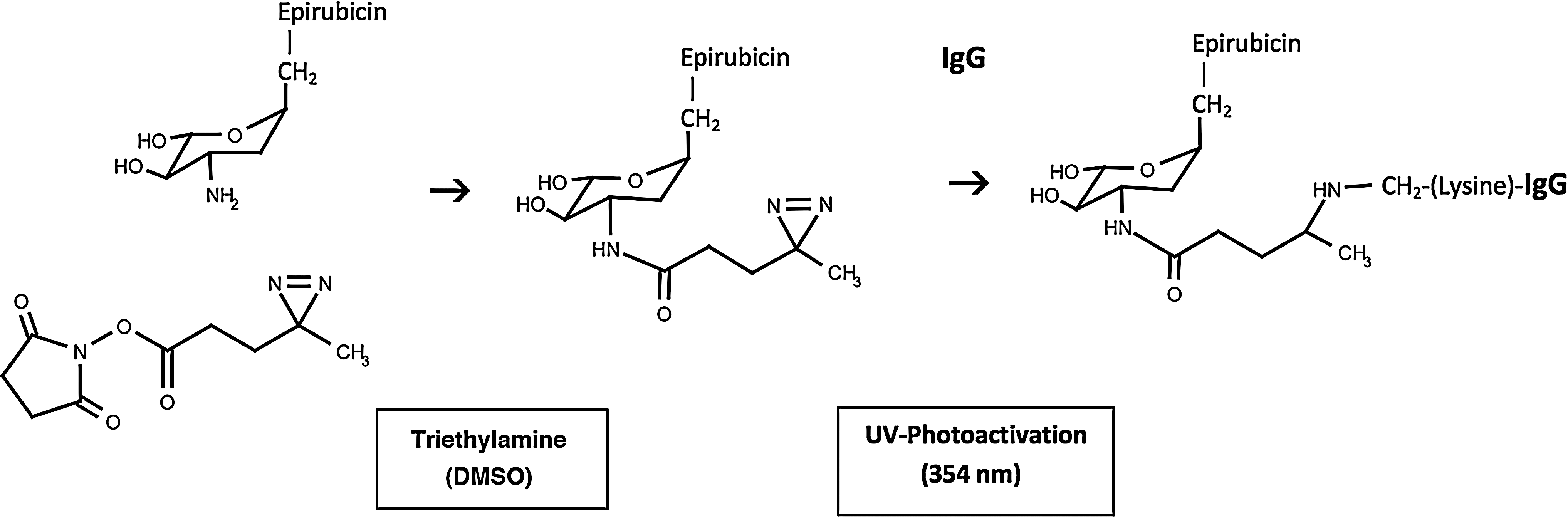
Schematic illustration of the chemical reactions involved in the synthesis of epirubicin-(C3-amide)-[anti-HER2/neu]. (Phase-I) creation of a covalent amide bond at the C3 monoamine of epirubicin and the ester group of succinimidyl 4,4-azipentanoate resulting in the creation of a covalent UV-photoactivated epirubicin-(C3-amide) intermediate accompanied by the liberation of the succinimide “leaving” complex; (Phase-II) creation of a covalent bond between the UV-photoactivated epirubicin-(C3-amide) intermediate and the ɛ-amine of lysine residues within the amino acid sequence of anti-HER2/neu monoclonal immunoglobulin initiated by photoactivation (UV 354 nm).
Phase-II synthesis scheme for covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic utilizing a UV-photoactivated epirubicin intermediate
Fractions of anti-HER2/neu monoclonal immunoglobulin (1.5 mg, 1.0×10−5 mmoles) in buffer (phosphate-buffered saline: phosphate 0.1, NaCl 0.15 M, EDTA 10 mM, pH 7.3) were combined at a 1:3.5 molar-ratio with the epirubicin (C3-amide) UV-photoactivated intermediate (Phase-1 end product) and allowed to gently mix by constant stirring for 5 minutes at 25°C in the dark. The photoactivated group of the epirubicin (C3-amide) intermediate was preferentially reacted with the ɛ-amine of lysine residues within the amino acid sequence of anti-HER2/neu monoclonal immunoglobulin during a 15 minute exposure to UV light at 354 nm (reagent activation range 320–370 nm) in combination with constant gentle stirring (Fig. 1). Residual epirubicin was removed from epirubicin-(C3-amide)-[anti-HER2/neu] applying micro-scale column chromatography with the media pre-equilibrated with PBS (phosphate 0.1, NaCl 0.15 M, pH 7.3).
Analysis, characteristics and properties
General analysis
Determination of the IgG concentration within covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic preparations was determined by measuring absorbance at 280 nm (Fig. 2). Measurement of epirubicin concentrations required excitation of epirubicin immunochemotherapeutic preparations at 485 nm followed by detection of emission at 538 nm. Known epirubicin reference controls were applied to generate a linear curve and the formulation of a mathematical equation that was utilized to accurately calculate and epirubicin concentrations between 10−9 and 10−5 M (final concentration). Determination of the concentration of nonconjugated “free” epirubicin contained in covalent epirubicin immunochemotherapeutic preparations was established by chloroform extraction, 60 –62 with the organic phase collected by pipette, evaporated to dryness under a stream of nitrogen gas, and the resulting residue dissolved in Tris buffered saline (TBS, 50 mM, pH 7.4) before quantitative analysis.

Molecular structure and chemical composition of the covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic created using a UV-photoactivated epirubicin intermediate that was preferentially bound covalently to the ɛ-amine of lysine residues within the amino acid sequence of anti-HER2/neu monoclonal immunoglobulin following exposure to UV-light (354 nm).
Nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis size separation, Western-blot immunodetection, and chemiluminescent autoradiography analyses
Standardized amounts and concentrations (60 μg/mL) of covalent epirubicin immunochemotherapeutic and reference control immunoglobulin fractions were combined 50/50 with an equal volume of conventional polyacrylamide gel electrophoresis (PAGE) sample preparation buffer (Tris/glycerol/bromphenyl blue/sodium dodecyl sulfate) formulated without 2-mercaptoethanol. Each immunoglobulin sample (0.9 μg/well) was processed without boiling and then developed in parallel with a mixture of prestained reference control molecular weight markers by nonreducing SDS-PAGE (11% acrylamide, 100 V constant voltage at 3°C for 2.5 hours). Developed nonreducing SDS-PAGE acrylamide gels were then equilibrated in electrophoresis “tank” buffer devoid of methanol. Lateral transfer of SDS-PAGE separated proteins onto sheets of nitrocellulose membrane for immunodetection (Western blot) analysis was performed at 20 volts constant voltage for 16 hours at 2°C to 3°C with the transfer manifold packed in crushed ice.
Nitrocellulose membranes with laterally transferred immunoglobulin fractions for immunodetection analysis and chemiluminescent imaging were equilibrated in TBS (Tris HCl 0.1 M, NaCl 150 mM, pH 7.5, 40 mL) at 4°C for 15 minutes followed by incubation in TBS blocking buffer solution (Tris 0.1 M, pH 7.4, 40 mL) containing bovine serum albumin (BSA 5%) applied at 2°C to 3°C for 16 hours in combination with gentle horizontal agitation. Before further processing nitrocellulose membranes were vigorously rinsed in TBS (Tris 0.1 M, pH 7.4, 40 mL, n=3 rinses).
Rinsed BSA-blocked nitrocellulose membranes for immunodetection (Western blot) analyses were then incubated with biotinylated goat anti-murine IgG (1:10,000 dilution) at 4°C for 18 hours applied in combination with gentle horizontal agitation. Following vigorous rinsing in TBS (pH 7.4, 4°C, 50 mL, n=3) nitrocellulose membranes were again incubated in BSA blocking buffer (BSA 5%, Tris 0.1 M, pH 7.4, 40 mL) at 4°C for 1 hour. Blocking buffer was decanted from nitrocellulose membrane blots and then vigorously rinsed in TBS (pH 7.4, 4°C, 50 mL, n=3 rinses) before incubation with strepavidin-[horseradish peroxidase] at 4°C for 2 hours applied in combination with gentle horizontal agitation (strepavidin-HRPO 1:100,000 dilution). Before chemiluminescent autoradiography nitrocellulose membranes were vigorously rinsed in TBS (Tris 0.1 M, pH 7.4, 40 mL, n=3 rinses). Chemiluminescent autoradiography next entailed incubation of nitrocellulose membranes in HRPO chemiluminescent substrate (25°C; 5 to 10 minutes) and the acquisition of images achieved by exposing radiographic film (Kodak BioMax XAR radiograph film) to nitrocellulose membranes sealed in transparent ultraclear resealable plastic bags.
Mammary carcinoma tissue culture cell culture
Human chemotherapeutic-resistant human mammary adenocarcinoma SKBr-3 was utilized as an ex vivo neoplasia model. Mammary adenocarcinoma SKBr-3 uniquely overexpresses EGFR1 (ErbB-1, HER1) and highly overexpresses EGFR2 (HER2/neu, ErbB-2, CD340, p185) at 2.2×105/cell and 1×106/cell respectively.
Propagation of mammary adenocarcinoma SKBr-3 was done in 150-cc 2 tissue culture flasks containing McCoy's 5a Medium Modified supplemented with fetal bovine serum (10% v/v) and penicillin-streptomycin at a temperature of 37°C under a gas atmosphere of air (95%) and carbon dioxide (CO2 5%). Tissue culture media was not supplemented with growth factors, growth hormones, or other biological stimulants of any type. Investigations were all performed using mammary adenocarcinoma SKBr-3 propagated to a ≥85% level of confluency.
Cell-ELISA IgG binding assay
Cell suspensions of mammary adenocarcinoma SKBr-3 were seeded into 96-well microtiter plates in aliquots of 2×105 cells/well and allowed to form confluent adherent monolayers over a period of 48 hours. The growth media within individual wells was then manually removed by pipette and the adherent mammary adenocarcinoma SKBr-3 monolayers serially rinsed (n=3) with PBS followed by stabilization on the plastic surface of microtiter plates with paraformaldehyde (4% in PBS, 15 minutes). Stabilized mammary adenocarcinoma SKBr-3 monolayers were then incubated with covalent epirubicin-(C3-amide)-[anti-HER2/neu] formulated at gradient concentrations in tissue culture growth media (0.010, 0.025, 0.050, 0.250, and 0.500 μg/mL IgG final well concentrations; 200 μL/well). Direct contact incubation of mammary adenocarcinoma SKBr-3 with epirubicin-(C3-amide)-[anti-HER2/neu] was performed at 37°C over a period of 3 hours under a gas atmosphere of air (95%) and carbon dioxide (CO2 5%). Residual non-cell bound epirubicin-(C3-amide)-[anti-HER2/neu] was removed by serial rinsing with PBS (n=3). Development of stabilized mammary adenocarcinoma SKBr-3 then entailed incubation with β-galactosidase-[goat anti-mouse IgG] (1:500 dilution) at 25°C for 20 hours followed by serial rinsing with PBS (n=3) to remove residual non-cell bound 2° immunoglobulin. In the final stages of cell-ELISA development stabilized mammary adenocarcinoma SKBr-3 monolayers were incubated with freshly formulated β-galactosidase substrate (nitrophenyl-β-D-galactopyranoside, 0.9 mg/mL in PBS pH 7.2 containing MgCl2 10 mM and 2-mercaptoethanol 0.1 M; 100 μL/well). Absorbance within each individual well was then measured at 410 nm after incubation at 37°C for a period of 15 minutes (630 nm reference wavelength).
Cell survival assay for measuring epirubicin-(C3-amide)-[anti-HER2/neu] cytotoxicity
Covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic was formulated in growth media at standardized epirubicin-equivalent concentrations of 10−10, 10−9, 10−8, 10−7, and 10−6 M (final well concentrations). Individual epirubicin-equivalent concentrations of covalent epirubicin-(C3-amide)-[anti-HER2/neu] were then transferred in triplicate into 96-well microtiter plates containing mammary adenocarcinoma SKBr-3 (200 μL/well).
Contents within individual 96-well microtiter plate wells were manually removed by pipette at 72 hours and then the mammary adenocarcinoma SKBr-3 monolayers were serially rinsed (n=3) with PBS followed by incubation with 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT formulated at 5 mg/mL in RPMI-1640 growth media devoid of pH indicator or bovine fetal calf serum). During a 3-hour incubation period at 37°C under a gas atmosphere of air (95%) and carbon dioxide (5% CO2) mammary adenocarcinoma SKBr-3 populations were allowed to biochemically convert intracellular MTT to navy-blue formazone crystals by endogenous mitochondrial succinate dehydrogenase. Contents of the 96-well microtiter plate were then manually removed by pipette followed by serially rinsing with PBS (n=3). The resulting navy blue intracellular formazone crystals were then dissolved with dimethyl sulfoxide (DMSO) (300 μL/well; 30 minutes, 25°C, continuous gentle horizontal agitation). Spectrophotometric absorbance was then measured at 570 nm for the blue-colored supernatants contained within in each well utilizing a computer-integrated microtiter plate reader.
Results
Synthetic chemistry
During Phase-I of the synthesis scheme the ester group of succinimidyl 4,4-azipentanoate was reacted with epirubicin resulting in the creation of a covalent amide bond at the C3 monoamine of the anthracycline carbohydrate moiety (daunosamine -NH2-3′) and liberation of a succinimide “leaving” complex (Fig. 1). The resulting Phase-I product was a covalent UV-photoactivated epirubicin-(C3-amide) intermediate that was preferentially reacted in Phase-II of the synthesis scheme with the ɛ-amine of lysine residues and other primary amines within the amino acid sequence of anti-HER2/neu immunoglobulin resulting in the creation of a covalent bond structure (Figs. 1 and 2). Epirubicin was formulated in molar excess of succinimidyl 4,4-azipentanoate to maximize production of the UV-photoactivated epirubicin-(C3 -amide) intermediate and minimize concentrations of residual unreacted reagents.
Molecular and physical properties
The percent of non-covalently bound anthracycline contained in epirubicin-(C3-amide)-[anti-HER2/neu] preparations following micro-scale desalting/buffer exchange column chromatography was consistently <4.0% where any residual non-covalently bound anthracycline remaining in the preparation is not available for removal by performing repeated/serial column chromatography separations. 63
The anthracycline molar-incorporation-index for epirubicin-(C3-amide)-[anti-HER2/neu] was 40% (39.65%). Related preliminary analyses revealed that the UV-photoactivated epirubicin-(C3-amide) intermediate in DMSO retained reactivity after freezing for at least 48 hours at −20°C based on an epirubicin molar-incorporation-index of 51% when the reaction mixture was combined with BSA at a [succinimidyl 4,4-azipentanoate]:BSA molar-ratio of 7:1 and 9:1 in concert with UV-photoactivation (354 nm) at 25°C for 15 minutes.
Evaluation of covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic following SDS-PAGE size separation by immunodetection (Western blot) analysis utilizing anti-murine IgG-strepavidin as an indicator 2° immunoglobulin produced chemiluminescent autoradiographic images that detected a single major immunoglobulin fraction of 150-kDa (Fig. 3). The profile of these images were highly analogous to the anti-HER2/neu reference control and similar to results previously reported for synthesis methodologies of other covalent immunochemotherapeutics. 2,7,40

Size-separation of covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in concert with immunodetection (Western-blot) analysis and chemiluminescent autoradiography. (Lane-1) murine anti-HER2/neu monoclonal immunoglobulin (reference control); and (Lane-2) covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic. Size-separated immunoglobulin preparations laterally transferred on sheets of nitrocellulose membrane were incubated with goat anti-mouse immunoglobulin-HRPO applied as a 1° immunoglobulin. Subsequent development entailed incubation of nitrocellulose membranes with an HRPO chemiluminescent substrate to facilitate acquisition of autoradiography images.
Cell-ELISA total membrane IgG binding analyses
Epirubicin-(C3-amide)-[anti-HER2/neu] formulated at standardized immunoglobulin-equivalent concentrations of 0.010, 0.025, 0.050, 0.250, and 0.500 μg/mL produced cell-ELISA profiles that detected proportional increases in total immunoglobulin bound to mammary adenocarcinoma SKBr-3 external surface membranes (Fig. 4).
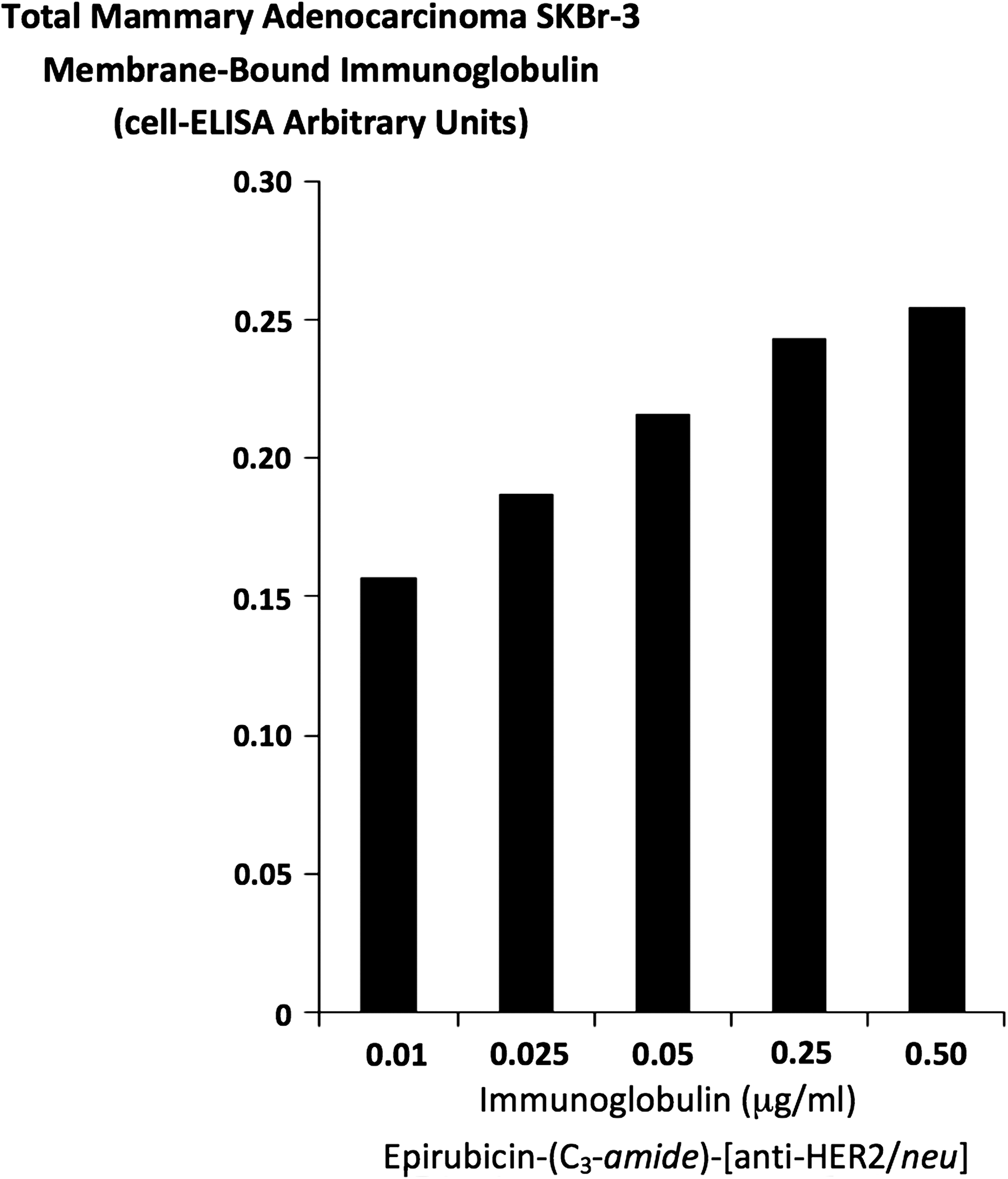
Detection of total immunoglobulin bound on the exterior surface membrane of mammary adenocarcinoma SKBr-3 monolayer populations as a function of increasing covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic concentration. Covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic. Monolayers of mammary adenocarcinoma SKBr-3 were incubated with covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic over a 4-hour period and then total immunoglobulin bound on the exterior surface membrane was detected by cell-ELISA analysis.
Cytotoxic anti-neoplastic potency
Maximum mean cytotoxic anti-neoplastic potency for covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic against chemotherapeutic-resistant mammary adenocarcinoma SKBr-3 was 88.5% (11.5% residual survival) at an epirubicin-equivalent concentration of 10−6 M (Fig. 5). Between final epirubicin-equivalent concentrations of 10−8 and 10−7 M there was a marked threshold increase in the mean cytotoxic anti-neoplastic potency of epirubicin-(C3-amide)-[anti-HER2/neu] from 9.9% to 66.9% (90.2% to 33.1% residual survival) (Fig. 5). Comparison of the mean cytotoxic anti-neoplastic activity profile for epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin alone against chemotherapeutic-resistant mammary adenocarcinoma SKBr-3 revealed a trend for the covalent anthracycline immunochemotherapeutic to exert slightly greater potency at epirubicin-equivalent concentrations between 10−10 and 10−6 μ but these differences were not statistically significant (Fig. 5). Mean cytotoxic anti-neoplastic potency profiles were highly analogous for [i] epirubicin-(C3-amide)-[anti-HER2/neu] synthesized utilizing a UV-photoactivated epirubicin-(C3-amide) intermediate; [ii] epirubicin-(C13-imino)-[anti-HER2/neu] synthesized using N-ɛ-maleimidocaproic acid hydrazide (EMCH) 26 ; and [iii] epirubicin-(C3-amide)-[anti-HER2/neu] with epirubicin-(C3-amide)-[anti-EGFR] formulated at 50/50 epirubicin-equivalent concentrations where each immunochemotherapeutic was synthesized using succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) 7 (Fig. 6). The anti-HER2/neu monoclonal immunoglobulin fraction alone did not exert any substantial anti-neoplastic potency against mammary carcinoma SKBr-3 at the end of a 72-hour incubation period which is in accord with previous investigations (Fig. 7). 7,26 –30,35,40
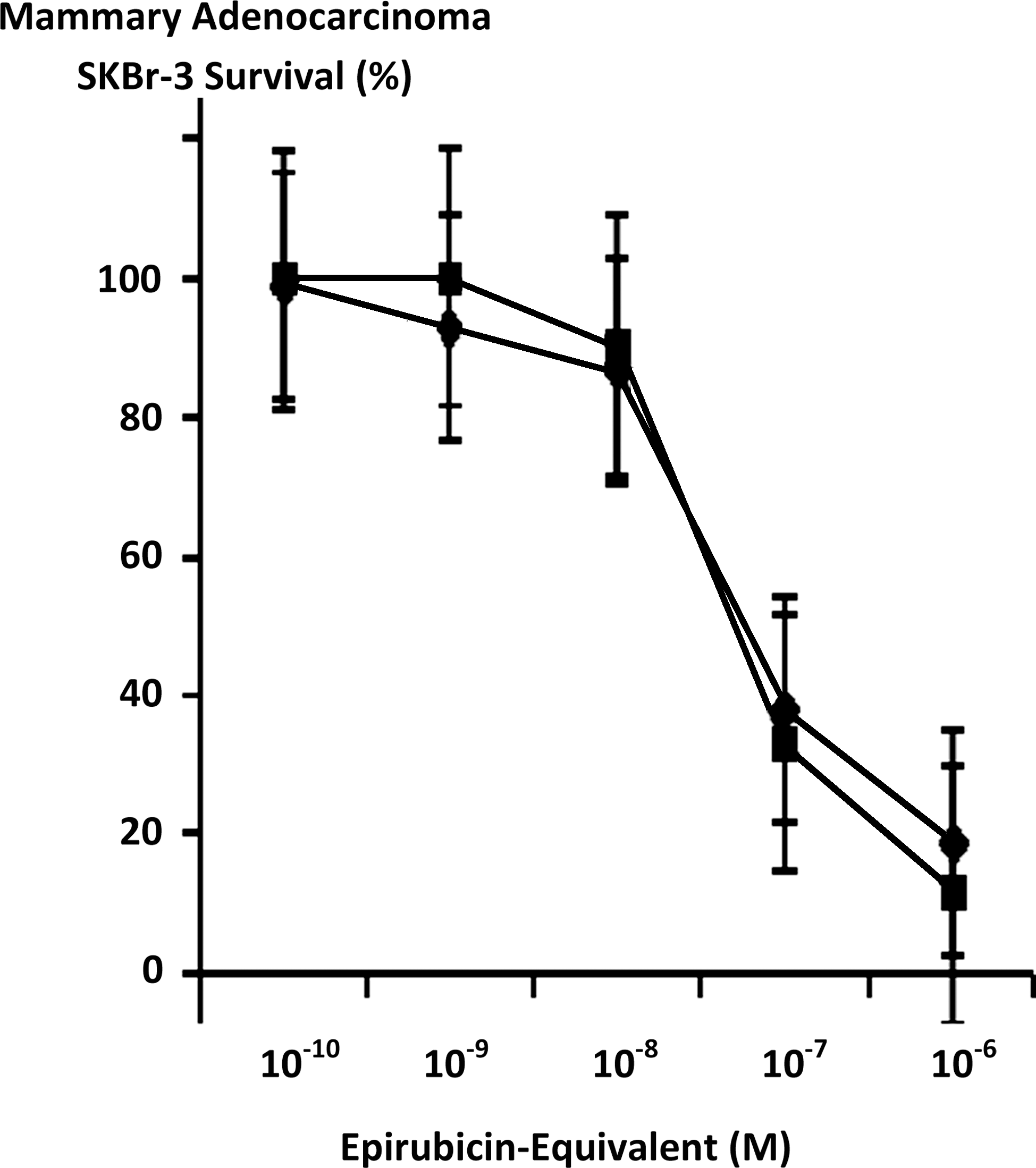
Influence of covalent bonding epirubicin to anti-HER2/neu monoclonal immunoglobulin based on the cytotoxic anti-neoplastic potency of epirubicin compared to epirubicin-(C3-amide)-[anti-HER2/neu] synthesized utilizing a UV-photoactivated epirubicin-(C3-amide) intermediate. (▪) covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic, and (♦) epirubicin chemotherapeutic. Mammary adenocarcinoma SKBr-3 monolayer populations were incubated with epirubicin or covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic for 72 hours and cytotoxic anti-neoplastic potency measured as a function of MTT cell vitality stain intensity relative to matched negative reference controls. MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide.
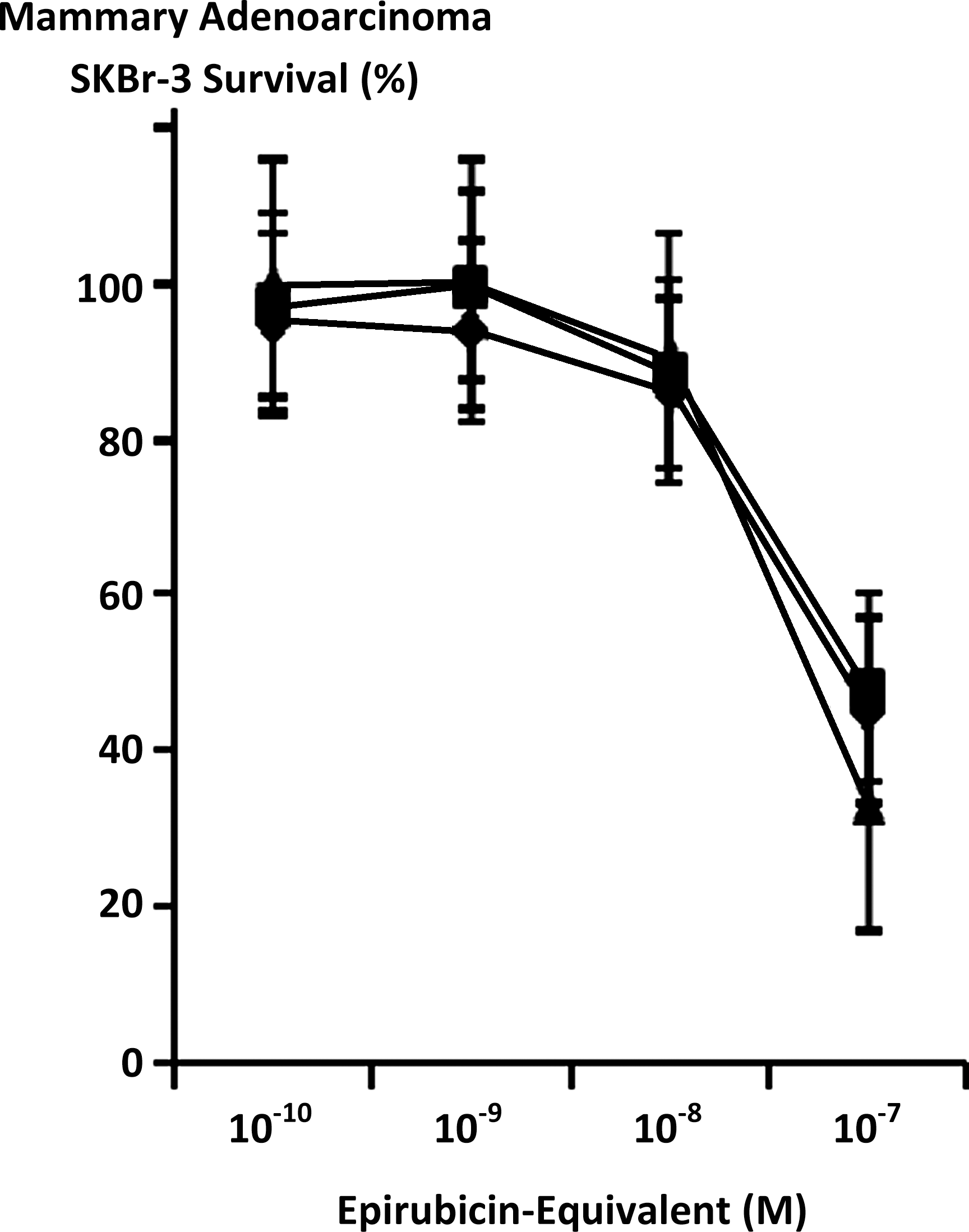
Relative cytotoxic anti-neoplastic potency of covalent epirubicin-[anti-HER2/neu] immunochemotherapeutics against chemotherapeutic-resistant mammary adenocarcinoma. (▴) epirubicin-(C3-amide)-[anti-HER2/neu] synthesized utilizing a UV-photoactivated epirubicin-(C3-amide) intermediate; (♦) epirubicin-(C13-imino)-[anti-HER2/neu] synthesized using N-ɛ-maleimidocaproic acid hydrazide (EMCH); and (▪) epirubicin-(C3-amide)-[anti-HER2/neu] with epirubicin-(C3-amide)-[anti-EGFR] as a 50/50 epirubicin-equivalent combination synthesized utilizing succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC). Mammary adenocarcinoma SKBr-3 monolayer populations were incubated with covalent epirubicin-immunochemotherapeutics for 72 hours and cytotoxic potency measured as a function of MTT cell vitality stain intensity relative to matched negative reference controls.
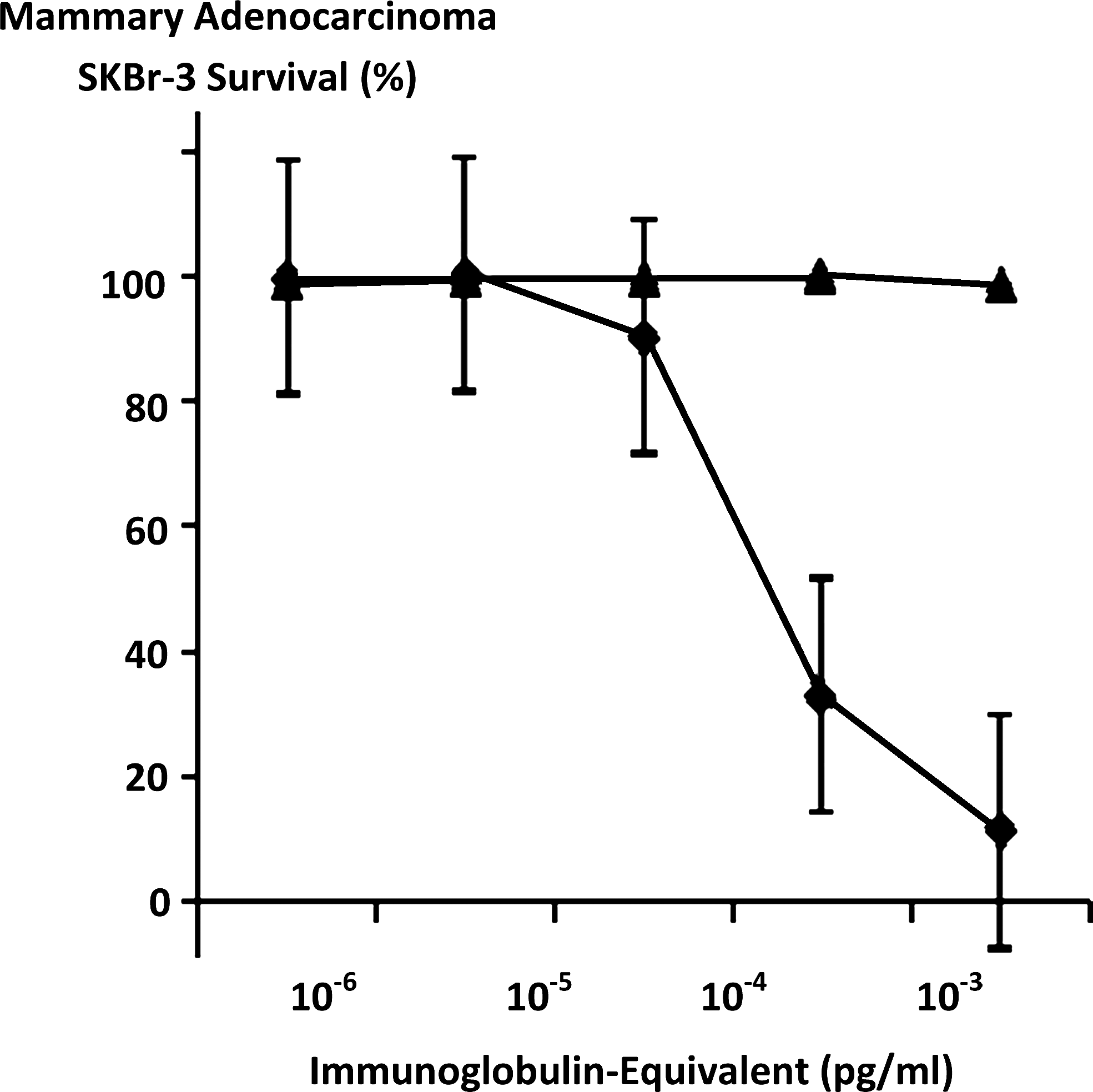
Relative anti-neoplastic potency of covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic synthesized utilizing a UV-photoactivated epirubicin-(C3-amide) intermediate compared to anti-HER2/neu monoclonal immunoglobulin against chemotherapeutic-resistant SKBr-3 mammary adenocarcinoma. (▴) covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic; and (♦) anti-HER2/neu monoclonal antibody. Monolayers of mammary adenocarcinoma SKBr-3 populations were incubated with the epirubicin immunochemotherapeutic and anti-HER2/neu monoclonal immunoglobulin fraction over a 72-hour period. Cytotoxicity anti-neoplastic potency was measured as a function of MTT cell vitality stain intensity relative to matched negative reference controls.
Discussion
A small spectrum of molecular platforms has been applied to facilitate selective “targeted” delivery of a variety of biological agents and conventional chemotherapeutics that can exert significant cytotoxic anti-neoplastic properties. Biological agents utilized in this regard include various immunotoxin preparations synthesized to enhance selective “targeted” delivery of Pseudomonas exotoxin, 64,65 cholera exotoxin, 66 diphtheria exotoxin, 67 ricin, 68,69 and genoline, 70 in addition to radioisotopes (e.g., [131I]-tositumomab, [177Lu]-octreotate, [90Yt-tiuxetan]-ibritumomab; [153Sm]-lexidronam, and [89Sr]-lexidronam). Chemotherapeutics that have been covalently bonded to molecular platforms for selective “targeted” delivery includes the anthracyclines, 7,26,35 gemcitabine, 30 methotrexate, 35,71 mitomycin, 35 the vinca alkaloids (modified analogs), 72 –74 bleomycine, 75,76 chlorambucil (non-IgG/transferrin), 77,78 cyclophosphamide, 79,80 paclitaxel (non-IgG), 81 –83 ozogamicin, 84,85 calicheamicins, 84 and monomethyl auristatin E. 86 –89
Several different chemical characteristics of the anthracycline class of chemotherapeutics can be utilized to develop multiple molecular designs and synthesis strategies allowing their covalent incorporation into immunoglobulin fractions or receptor ligands applying a variety of organic chemistry reactions. One method entails the reaction of both the carbohydrate C3 monoamine group of anthracyclines and the ɛ-amine of lysine residues within the amino acid sequence of immunoglobulin with the aldehyde groups found in sodium periodate oxidized dextran. 39 –41,90,91 Regulating the rate and extent of these types of synthesis reactions so that a largely homogenous product is generated is challenging because oxidized dextran functions as a somewhat nonselective homobifunctional covalent cross-linking agent. Generation of extraneous “side products” may in part explain the low potency occasionally reported for doxorubicin-dextran-immunoglobulin prepared using dextran as a molecular bridge. 39 –41,90,91 Polyethylene glycol has also been used as a similar synthetic bridge between immunoglobulin and doxorubicin, 92 A semi-synthetic scheme that is chemically analogous to the oxidized-dextran conjugation method utilizes the enzymatic coupling of anthracyclines at their α-monoamine terminus with the aldehydes of oxidized galactose residues of immunoglobulin yielding a Schiff base that is subsequently stabilized by mild reduction with pyridine borane. 43 A second conjugation method used for synthesizing a covalent bond between anthracyclines and a large molecule utilizes glutaraldehyde as homobifunctional cross-linking reagent that forms a “bridge” through covalent bond formation at the C3 monoamine of the anthracycline carbohydrate moiety (daunosamine -NH2-3′). 40 Production of doxorubicin-immunochemotherapeutics with glutaraldehyde however, can lead to excessive generation of chemotherapeutic/immunoglobulin precipitates and substantial declines in final product immunoreactivity. 40 Covalent doxorubicin-immunochemotherapeutics 27,40 and covalent doxorubicin-receptor fragment complexes 93 have been successfully produced using this synthesis scheme. Reactions of this type, however, need to be closely controlled because the resulting covalent anthracycline C3-amide immunoconjugate derivative can often be simultaneously created along with excessive immunoglobulin polymerization and extraneous side reactions if reagent formulations are less than ideal.
Covalent bond forming reactions have been devised with cross-linking reagents other than oxidized dextran (aldehyde groups) and glutaraldehyde that create covalent acid-labile bonds at the C3 monoamine found on the anthracycline carbohydrate moiety (daunosamine -NH2-3′). The covalent bond-forming reagent, cis-aconitic anhydride 29,34,40,61,94 –96 promotes covalent acylation at the C3 monoamine group found within the anthracycline carbohydrate moiety (daunosamine -NH2-3′) whereby it creates cis- and trans-isomers of N-cis-aconityl-anthracycline. 37,40,61,95,97 In practice, cis-aconitic anhydride has been used to synthesize both doxobucin 29,35 and daunorubicin 34,35 covalent immunochemotherapeutics. Similar to dextran and glutaraldehyde, cis-aconitic anhydride forms covalent bonds with immunoglobulin fractions at the ɛ-amine group (R-NH2) of lysine amino acid residues 97 but is associated with several of the same disadvantages experienced with many other homobifunctional cross-linking agents.
In addition to semi-synthetic methodologies that introduce molecular cross-linking agents at the C3 monoamine group found within the anthracycline carbohydrate moiety (daunosamine -NH2-3′), several strategies have been developed that create a covalent bond at the C13 keto (RC(=O)R′ position chemotherapeutic class. A versatile method of producing anthracycline-immunoconjugates utilizes the organic polymer, N-(2-hydroxypropyl)methacrylamide (HPMA) whereby doxorubicin is cross-linked with HPMA through formation of a hydrazone bond at the chemotherapeutic C13 keto (RC(=O)R′ position, 98 or a N-cis-aconityl reaction at the C3 monoamine group of anthracyclines. 60,61 The resulting anthracycline-HPMA analog can then be conjugated with sodium periodate oxidized, or N-succinimidyl 3-(2-pyridyldithio)-propionate modified immunoglobulin fractions.
The C13 keto (RC(=O)R′ of the anthracyclines can be reacted with a hydrazide 3,60,99 –101 to create a hydrazone covalent bond. 3,26,44,61,98,102 –104 Hydrazides like 4-[N-maleimidomethyl]cyclohexane-1-carboxyl-hydrazide, 28,32 6-maleimidocaproyl-hydrazide (3,3′-N-[ɛ-maleimidocaproic acid] hydrazide), 2 –4,31,100,103 N-(2-hydroxypropyl)methacrylamide (HPMAO) based analogs, 60,105 EMCH, 26 and other maleimido-hydrazides 3 have been used in this capacity. Maleimido groups are commonly used in combination with hydrazides resulting the creation of sulfhydryl-reactive hydrazone-linked maleimido-anthracycline intermediates (e.g., 6-maleimidocaproyl)-hydrazone-doxorubicin) that is subsequently reacted with cystine amino acid residues, 2,3,7,25,26 or sulfhydryl-modified (thiolated) ɛ-amine of lysine residues within the amino acid sequence of immunoglobulin, 28,31,60,99,106 albumin, 2,3 or other biological protein fractions.
Synthetic chemistry reactions for covalent epirubicin-(C3-amide)-[anti-HER2/neu]
The synthesis of covalent epirubicin-(C3-amide)-[anti-HER2/neu] utilizing succinimidyl 4,4-azipentanoate initially involved reacting its ester group with the C3 monoamine of the anthracycline carbohydrate moiety (daunosamine -NH2-3′). The resulting UV-photoactivated epirubicin-(C3-amide) intermediate was then utilized to create a covalent bond at the ɛ-amine of lysine residues within the amino acid sequence of anti-HER2/neu immunoglobulin initiated by brief UV photoactivation. The epirubicin-(anti-HER2/neu) molar-incorporation-index of 40% (39.65%) was comparable to analogous levels calculated when using the heterobifunctional reactants, succinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (SMCC), 7 EMCH, 26 or N-[p-maleimidophenyl]-isocyanate (PMPI). 30 In contrast to SMCC, 7,107 –109 EMCH, 9,10,26 and PMPI, 30,110 –112 succinimidyl 4,4-azipentanoate is technically a homobifunctional reactant when it is applied under two separate conditions that both promoted covalent bond formation at primary amine groups. In this context, the NHS-ester group creates a covalent bond with amines in aqueous buffer or organic solvent systems in the presences of a proton donor (triethylamine) while the photoactivated diazirine group forms covalent bonds with amines in response to UV light exposure. One important implication of this difference is that when a chemotherapeutic with an amine group is reacted in molar excess with succinimidyl 4,4-azipentanoate during Phase-I of the synthesis scheme there is still a relatively large amount of residual unreacted chemotherapeutic remaining in solution. In effect, this large amount of residual unreacted chemotherapeutic competes with the ɛ-amine of lysine residues within the amino acid sequence of immunoglobulin during Phase-II UV photoactivation. Given this concept, the synthesis scheme can be further optimized to improve Phase II epirubicin molar-incorporation-indexes if the anthracycline -to- succinimidyl 4,4-azipentanoate molar-ratio employed during the Phase-I synthesis reaction is reduced (e.g., 81% epirubicin incorporation index with a 1.5:1 molar-ratio of epirubicin -to- succinimidyl 4,4-azipentanoate).
Synthesis of epirubicin-(C3-amide)-[anti-HER2/neu] utilizing succinimidyl 4,4-azipentanoate does not require the creation or introduction of reduced sulfhydryl groups into the amino acid sequence of whole immunoglobulin, F(ab’)2, Fab, or other biologically active proteins. The lack of a requirement for protein thiolation is in obvious contrast to most other synthesis regimens that utilize heterobifunctional reactants similar to SMCC, 7,107 –109 EMCH, 9,10,26 or PMPI 30,110 –112 that contain a sulfhydryl-reactive moiety.
Pre-thiolation of immunoglobulin, F(ab’)2, Fab, or other biological proteins is usually required due to the relatively low number of nonsterically hindered sulfhydryl groups in the form of reduced cysteine amino acid residues (e.g., R-SH) available within their amino acid sequence. Increasing the number of available reduced sulfhydryl groups can be achieved by the application of 1,4-dithiothreitol which reduces intramolecular cystine-cystine bonds 28,31,32 and similar disulfide structures 113 (DTT: R-CH2-S-S-CH2-R → 2 R-CH2-SH). The actual synthetic introduction of “new” or additional reduced sulfhydryl groups at the ɛ-amine of lysine amino acid residues is possible with reactions that utilize 2-iminothiolane (2-IT), 2,6,26,30,114 mercaptosuccinimide, 115 or N-succinimidyl-S-acetylthioacetate (SATA). 7,114,116 Alternatively, carboxyl groups on molecules like heparin and hyaluronic acid can be thiolated with 3,3′dithiobis(propanoic)-hydrazide 113,117 or divinylsulfone, 92,118 for hydroxyl groups of molecules with a cholesterol-like core. 119 In the application of DTPH the integral disulfide bond is subsequently reduced with DTT reagent. 113,117
Covalently bonding epirubicin or other chemotherapeutic agents to biological protein fractions like immunoglobulin without a requirement for converting existing cystine-cystine disulfide bonds to their reduced form or the synthetic introduction of reduced sulfhydryl groups is a significant advantage not extensively described to date. Such synthesis schemes entail the implementation few synthetic chemistry reactions, require smaller amounts of critical reagents, and maximize final yield in part due to at least one less column chromatography separation procedure. The brief duration of the synthesis scheme for epirubicin-(C3-amide)-[anti-HER2/neu] utilizing succinimidyl 4,4-azipentanoate is also possible because of the relatively rapid time course for Phase-I and especially the Phase-II organic chemistry reactions, and because the methodology has been designed so that adjustment of buffer pH to different levels during the procedure is not necessary in contrast to other techniques. 97
Perhaps one of the most important features of the synthesis methodology is a lack of a requirement for cystine-cystine disulfide bond reduction or pre-thiolation which by design allows the application of synthetic chemistry reactions that are highly efficient under relatively milder conditions and promote a lower risk of protein fragmentation or secondary polymerization through premature intermolecular or intramolecular disulfide bond formation. 2 Realized benefits therefore include greater retained biological activity (e.g., antigen binding-avidity) and total final yield. Lastly, lack of a requirement for either converting existing cystine-cystine disulfide bonds to their reduced form or the introduction of reduced sulfhydryl groups into immunoglobulin fractions reduces restrictions and limitations on the magnitude of the molar-incorporation-index that can be attained. In contrast, the chemotherapeutic incorporation index for covalent immunochemotherapeutics synthesized utilizing SMCC, 7,107 –109 EMCH, 9,10,26 or PMPI 30,110 –112 is limited or restricted to levels only as high or lower than the extent of lysine ɛ-amine prethiolation. In pre-thiolation dependent synthesis schemes higher epirubicin molar-incorporation-indexes are possible with modifications in methodology but the resulting harsher synthesis conditions are accompanied by substantial reductions in total yield of covalent immunochemotherapeutic, 6 and declines in antigen-immunoglobulin binding-avidity (e.g., cell-ELISA parameters).
The implementation of succinimidyl 4,4-azipentanoate in the synthesis scheme for epirubicin-(C3-amide)-[anti-HER2/neu] has other desirable attributes besides a lack of requirement for immunoglobulin pre-thiolation. In contrast to SMCC, 7,107 –109 EMCH, 9,10,26 or PMPI 30,110 –112 the synthesis of epirubicin-(C3-amide)-[anti-HER2/neu] utilizing succinimidyl 4,4-azipentanoate has the added benefit of not introducing biologically irrelevant five and six carbon or carbon/nitrogen ring structures into the final form of covalent immunochemotherapeutics (Figs. 1 and 2). Elimination of any extraneous ring structures decreases the probability of inducing an in vivo humoral immune response when administered by IV injection that can ultimately result in the formation of neutralizing antibody and an increased risk of a post-treatment immune hypersensitivity reaction. The Phase-I reaction can be performed either in an aqueous buffer or in an organic solvent system containing a low concentration of triethylamine [N(CH2CH3)3] or other proton acceptor molecule. In stock solutions or reaction mixtures that contain an aqueous buffer solution significant hydrolytic degradation of succinimidyl 4,4-azipentanoate occurs. Alternatively, if stock solutions and reaction mixtures of epirubicin with succinimidyl 4,4-azipentanoate are instead formulated in an anhydrous organic solvent like DMSO in combination with a proton acceptor molecule then the resulting UV-photoactivated epirubicin-(C3-amide) intermediate is stable at 4°C or −20°C for a period of time when adequately protected from UV-light exposure. Such properties for succinimidyl 4,4-azipentanoate further demonstrate the convenient options of the synthesis method described that are in part facilitated by the ability to “presynthesize” and store the UV-photoactivated epirubicin-(C3-amide) intermediate for an extended period for use in the future production of covalent epirubicin-immunochemotherapeutics. The design of the synthesis scheme described offers still another added level of convenience because it illustrates a model method that can be adapted and modified to facilitate the covalent bonding of an array of different chemotherapeutic agents to a wide range of immunoglobulins (e.g., IgG, Fab’), receptor ligands, or similar biologically active protein fractions.
Molecular and physical properties
Size separation by SDS-PAGE was used in concert with lateral transfer on nitrocellulose membrane, immunodetection analyses, and chemiluminescent autoradiography to validate the absence of any detectable IgG-IgG polymers or peptide fragmentation of the anti-HER2/neu component within epirubicin-(C3-amide)-[anti-HER2/neu] preparations (Fig. 3). Collectively the lack of detectable IgG-IgG polymerization or IgG fragmentation directly correlates with the high level of retained antigen binding-avidity for anti-HER2/neu within the preparation of covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic.
Cell-ELISA membrane IgG binding analysis
Total membrane bound IgG profiles in mammary adenocarcinoma SKBr-3 for epirubicin-(C3-amide)-[anti-HER2/neu] synthesized utilizing succinimidyl 4,4-azipentanoate proportionately increased with elevations in total immunoglobulin concentration (Fig. 3). The total amount of epirubicin-(C3-amide)-[anti-HER2/neu] synthesized with succinimidyl 4,4-azipentanoate that was bound to the external surface membrane of mammary adenocarcinoma SKBr-3 was relatively higher than that previously observed for epirubicin-(C3-amide)-[anti-HER2/neu] synthesized with SMCC in combination with SATA pre-thiolation 7 or epirubicin-(C13-imino)-[anti-HER2/neu] synthesized with EMCH in combination with 2-IT pre-thiolation 26 or gemcitabine-(C5-carbamate)-[anti-HER2/neu] synthesized with PMPI in combination with 2-IT pre-thiolation. 30 Presumably the difference reflects a higher degree of binding-avidity (biological integrity) of epirubicin-(C3-amide)-[anti-HER2/neu] for exterior surface membrane associated HER2/neu because succinimidyl 4,4-azipentanoate does not require pre-thiolation of immunoglobulin or other biological protein fractions. Prethiolation under certain circumstances can result in premature formation of intramolecular and intermolecular disulfide bond structures. Binding-avidity of epirubicin-(C3-amide)-[anti-HER2/neu] for membrane HER2/neu is also dependent upon and modified by the extent that a chemotherapeutic is covalently introduced within the Fab’ antigen-binding region of anti-HER2/neu immunoglobulin. In this context, seemingly modest alterations in synthesis chemistry and elevations in chemotherapeutic molar-incorporation-index can profoundly influence immunoglobulin binding properties. 29 Given this perspective, the relatively mild conditions employed during the course of synthesis procedures and relatively modest molar-incorporation-index of 40.0% contributed to the high biological integrity of epirubicin (C3-amide)-[anti-HER2/neu] based on results from SDS-PAGE/immunodetection/chemiluminescent autoradiography and cell-ELISA analyses.
Detection of measurable increases in epirubicin-(C3-amide)-[anti-HER2/neu] binding to surface membrane HER2/neu complexes as a function of increases in the concentration of covalent epirubicin-immunochemotherapeutic is critically important from the perspective of validating retained biological integrity and quality control assurances. Implementing synthesis schemes that aid in maintaining maximum epirubicin-(C3-amide)-[anti-HER2/neu] binding-avidity for surface membrane HER2/neu serves to optimize the magnitude of selective “targeted” epirubicin delivery at HER2/neu trophic receptors highly overexpressed by mammary adenocarcinoma SKBr-3 populations. Second, maintaining maximum binding-avidity also directly facilitates efficient and more extensive internalization of epirubicin-(C3-amide)-[anti-HER2/neu] by mechanisms of ligand-induced receptor-mediated-endocytosis at HER2/neu overexpressed by mammary adenocarcinoma 7,26,30,120 and other neoplastic cell types. 121 –125 Seemingly modest alterations in synthetic chemistry conditions, and elevations in chemotherapeutic molar-incorporation-indexes can profoundly influence the selective binding properties of immunoglobulin fractions. 29 Relatively higher anthracycline-immunoglobulin molar-incorporation-indexes could have been attained during the synthesis of epirubicin-(C3-amide)-[anti-HER2/neu] utilizing succinimidyl 4,4-azipentanoate. However, such attributes would have been attainable with modifications that have the potential to promote substantial declines in final product yield and antigen binding-avidity secondary to the requisite application of harsher conditions during synthetic reactions. In this regard, modifications in synthesis scheme conditions that result in higher chemotherapeutic incorporation ratios may also only result in modest declines in immunoreactivity (e.g., −14% for a 73:1 ratio) but disproportionately large declines in anti-neoplastic potency in vivo down to levels that are substantially lower than those found with nonconjugated “free” anthracycline. 39
Cytotoxic anti-neoplastic potency
The cytotoxic anti-neoplastic activity of covalent epirubicin-(C3-amide)-[anti-HER2/neu] immunochemotherapeutic following binding to HER2/neu trophic receptors expressed by mammary adenocarcinoma SKBr-37,26,30 likely occurs through mechanisms of receptor-mediated-endocytosis analogous to that previously described for similar covalent anthracycline immunochemotherapeutics. 121 Although specific kinetic data for the tropic receptors HER2/neu and EGFR expressed by mammary adenocarcinoma is limited metastatic multiple myeloma is known to internalize and metabolize ∼8×106 molecules of anti-CD74 monoclonal antibody per day. 126
The profile for the cytotoxic neoplastic potency of epirubicin-(C3-amide)-[anti-HER2/neu] synthesized with succinimidyl 4,4-azipentanoate was highly analogous to chemotherapeutic-equivalent concentrations of [i] epirubicin, in addition to levels measured for [ii] epirubicin-(C13-imino)-[anti-HER2/neu] synthesized with EMCH 26 ; and [iii] epirubicin-(C3-amide)-[anti-HER2/neu] in combination with epirubicin-(C3-amide)-[anti-EGFR] whereby both covalent immunochemotherapeutics were synthesized utilizing SMCC 7 (Figs. 4 and 6). Such findings are also analogous to the greater 27,29,35,42,43,101,102,127 or relatively high (effective) 39 levels of cytotoxic anti-neoplastic potency noted for similar covalent anthracycline-immunochemotherapeutics synthesized under optimal conditions when evaluated against other neoplastic cell types applying standardized anthracycline-equivalent concentrations.
The essentially comparable level of cytotoxic anti-neoplastic potency for epirubicin-(C3-amide)-[anti-HER2/neu] synthesized using succinimidyl 4,4-azipentanoate compared to molar equivalent concentrations of epirubicin is collectively attributed to mechanisms of selective membrane deposition, continual receptor-mediated-endocytosis that in turn facilitates progressive intracellular internalization and cytosol accumulation of epirubicin. 7,26,29,30,43,121,128 Receptor-mediated-endocytosis at membrane-associated HER2/neu complexes in this fashion can promote elevations in intracellular chemotherapeutic concentrations that approach and exceed 8.5× 43 to >100× fold greater 128 levels than are possible through processes of simple passive anthracycline diffusion. In this scenario, >50% of selectively delivered anthracycline at 24 hours is intracellularly retained 43 where it becomes primarily associated with either membrane structures or it distributes more extensively throughout the cytosol environment. 121,129 Conversely, “free” anthracycline following passive diffusion across the intact lipid bilayer of cell membranes is primarily detected complexed with nuclear DNA <30 minutes after initial exposure 121 while anthracycline liberated from covalent anthracycline-immunochemotherapeutics reportedly distributes to, and accumulates within the nucleus, mitochondria, and golgi compartments. 63
One of the primary objectives for the molecular design, synthesis, and evaluation of the cytotoxic potency of [i] epirubicin-(C3-amide)-[anti-HER2/neu] synthesized with succinimidyl 4,4-azipentanoate (Figs. 1 and 2); [ii] epirubicin-(C13-imino)-[anti-HER2/neu] synthesized with EMCH 26 ; [iii] epirubicin-(C3-amide)-[anti-HER2/neu] and epirubicin-(C3-amide)-[anti-EGFR] both synthesized utilizing SMCC 7 ; and [iv] gemcitabine-(C5-carbamate)-[anti-HER2/neu] synthesized with PMPI 30 was to develop working prototypes of different covalent immunochemotherapeutics. Several variables could have been modified to achieve higher cytotoxic levels of anti-neoplastic potency for epirubicin-(C3-amide)-[anti-HER2/neu] at lower epirubicin-equivalent concentrations. First, the length of time that epirubicin-(C3-amide)-[anti-HER2/neu] was incubated in direct contact with mammary adenocarcinoma SBKr-3 could have been extended beyond 72 hours. 21,30,130,131 In doing so, mammary adenocarcinoma SKBr-3 is given an extended opportunity to continually internalize progressively more epirubicin-(C3-amide)-[anti-HER2/neu] by mechanisms of receptor-mediated-endocytosis.
Second, cytotoxic anti-neoplastic potency could have been measured utilizing a neoplastic cell type that is not generally classified as being chemotherapeutic-resistant analogous to many if not majority of the reports published in the literature to date (Figs. 4 –6). Few anthracycline-immunoconjugates have been reported to exert relatively greater cytotoxic anti-neoplastic potency against chemotherapeutic (multidrug) resistant neoplastic cell populations compared with the same anthracycline chemotherapeutic in “free” unbound form. 35 Exceptions include [i] covalent epirubicin-(C3-amide)-[anti-HER2/neu], 7 epirubicin-(C3-amide)-[anti-EGFR], 7 and epirubicin-(C13-imino)-[anti-HER2/neu], 26 immunochemotherapeutics (chemotherapeutic-resistant mammary adenocarcinoma SKBr-3); [ii] covalent anthracycline ligand-chemotherapeutics utilizing EGF or an EDF fragment (chemotherapeutic-resistant mammary carcinoma MCF-7AdrR) 93 ; and [iii] covalent daunorubicin immunochemotherapeutic synthesized using anti-chondroitin sulfate proteoglycan 9.2.27 surface marker (chemotherapeutic-resistant metastatic melanoma M21). 29,35,132
Third, early-stage cellular injury instead of cytotoxic anti-neoplastic potency could have been measured based upon either the detection of declines in cellular [H3]-thymidine incorporation (proliferation analysis), or alterations in ATP utilization using an ATP-dependent assay methodology because each of these analysis methodologies is reportedly ≥10× fold more sensitive in detecting early cell injury than MTT reagent based cell vitality assays. 133,134 Despite this consideration, assays that utilize MTT or similar cell vitality stain reagents for measuring cellular proliferation and growth continue to be extensively applied for the routine assessment of true chemotherapeutic cytotoxic anti-neoplastic potency. 7,26,30,135 –139
Fourth, cytotoxic anti-neoplastic potency could have been delineated in vivo against human neoplastic xenographs in animal hosts as a model for human neoplasia where the efficacy of a covalent immunochemotherapeutic frequently tends to be higher than in ex vivo tissue culture based models utilizing the same identical cancer cell type. 39,42,140 The enhanced levels of covalent immunochemotherapeutic potency measured in vivo is presumed in part to be dependent upon contributions from endogenous immune responses that include antibody-dependent cell cytotoxicity (ADCC) phenomenon in concert with complemented-mediated cytolysis initiated by the formation of antigen-immunoglobulin complexes on the exterior surface membrane of “targeted” neoplastic cells. During ADCC events, the cell types that are actively involved in this response release cytotoxic components that are known to additively and synergistically enhance the cytotoxic anti-neoplastic activity of conventional chemotherapeutic agents. 141 Contributions of ADCC and complement-mediated cytolysis to the in vivo cytotoxic anti-neoplastic potency of covalent anthracycline immunochemotherapeutics would be further complemented by, and directly correlate with the additive and synergistic levels of anti-neoplastic potency produced by anti-trophic receptor monoclonal immunoglobulin applied in dual combination with nonconjugated chemotherapeutic agents. 48 –50,53 –59,113,118 Additive or synergistic interactions of this type have been detected with anti-HER2/neu in concert with cyclophosphamide, 48,49 docetaxel, 48 doxorubicin, 48,49 etoposide, 48 methotrexate, 48 paclitaxel, 48,49 or vinblastine. 48
Fifth, several modifications could have been made in the synthesis strategy in order to increase the anthracycline molar-incorporation index. Relevant examples in this regard include the application of the anthracycline and covalent bond forming reagent at higher concentrations; implementation of smaller reaction volumes during the synthesis procedures; increase the duration of Phase I and Phase II synthesis schemes; and possibly altering the molar ratios of the anthracycline, covalent bond forming reagent and immunoglobulin in a manner that forces each reaction in a direction that increases the final product yield. Unfortunately, as previously eluded to, such changes, frequently require employing harsher synthesis conditions and a higher risk of reduced biological function (e.g., antigen-immunoglobulin binding-avidity) and substantial declines in product yield. 6,39 In the preparation of some covalent immunochemotherapeutics, relatively higher anthracycline-immunoglobulin molar-incorporation-indexes results in only modest declines in immunoreactivity (e.g., 86% for a 73:1 ratio) but disproportionate declines in anti-neoplastic activity down to potency levels substantially lower than those found with nonconjugated “free” anthracycline. 39 However, in contrast to succinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate (SMCC), 7,107 –109 EMCH, 8 –10,26,142,143 or PMPI 30,110 –112 the utilization of succinimidyl 4,4-azipentanoate allows greater flexibility for increasing the chemotherapeutic molar-incorporation-index while synthesizing covalent immunochemotherapeutics without the simultaneous creation of harsher reaction conditions. Therefore the major risk of compromising the biological integrity (antigen binding avidity) of epirubicin-(C3-amide)-[anti-HER2/neu] synthesized with succinimidyl 4,4-azipentanoate is almost entirely associated with introducing too many chemotherapeutic moieties into the Fab antigen bindings regions of the immunoglobulin molecule. Despite the considerations related to chemotherapeutic molar-incorporation-index, it should be emphasized that mathematically it appears that the expression density of external membrane-associated “targets” used to facilitate selectively “targeted” chemotherapeutic delivery is a critically most important variable that significantly determines cytotoxic anti-neoplastic potency. In this regard, it is important that external membrane-associated sites be chosen that are known to functionally undergo phenomenon analogous to receptor-mediated-endocytosis to avoid the simple “coating” of cancer cell external surface membranes with a covalent immunochemotherapeutic. Such a prerequisite assumes that the chemotherapeutic agent applied for the synthesis of a covalent immunochemotherapeutic has a mechanism-of-action that is dependent upon entry into the cytosol or nucleus in contrast to agents that alter the physical integrity of neoplastic cell external membranes.
Footnotes
Acknowledgments
Investigations were supported with indirect costs allocated from a grant that were awarded for an unrelated field of research. The authors would like to thank Mr. Tom Thompson in the Office of Agriculture Communications for assistance with photographing the autoradiography illustration. Professional consultants were not employed during the course of investigation and no individual received an honoraria or stock options.
Disclosure Statement
The authors claim they do not have an institutional or collaborative research arrangement with a commercial industry that might pose as a conflict of interest regarding the publication of research findings described in this manuscript.