
Abstract
Several F-18-labeled 2-nitroimidazole (azomycin) derivatives have been proposed for imaging hypoxia using positron emission tomography (PET). Their cell penetration is based on passive diffusion, which limits their intracellular concentration maxima. The purpose of this study was to investigate the uptake of N-(2-[18F]fluoro-3-(6-O-glucosyl)propyl-azomycin ([18F]F-GAZ), a new azomycin-glucose conjugate, in vitro and in vivo. [18F]F-GAZ was synthesized from its tetraacetyl nosylate precursor by nucleophilic radiofluorination. [18F]F-GAZ was evaluated in vivo in EMT-6 tumor-bearing Balb/C mice utilizing the PET and biodistribution analysis. In vitro uptake of [18F]FDG by EMT-6 cells was measured in the presence of unlabeled F-GAZ, 2-FDG, and D-glucose. [18F]F-GAZ was rapidly cleared from all tissues, including the blood pool and kidneys, with ultimate accumulation in the urinary bladder. Uptake of tracer doses of [18F]F-GAZ into EMT-6 tumors was fast, reaching a standardized uptake value of 0.66±0.05 within 5–6 minutes postinjection (p.i.), and decreased to 0.24±0.04 by 60 minutes p.i. (n=6). A tumor–muscle ratio of 1.87±0.18 was observed after 60 minutes. Total uptake of [18F]F-GAZ in tumors (60 minutes) amounted to 1.25%±0.15% ID/g versus 0.61%±0.14% ID/g (n=4) in muscle. Similar biodistribution and excretion were observed using carrier-added (100 mg/kg) doses of F-GAZ. In vitro, D-glucose and unlabeled 2-FDG were two orders of magnitude more potent than F-GAZ as competitive inhibitors of [18F]FDG uptake into EMT-6 cells. Besides its interaction with glucose transporters, F-GAZ seems to be not transported in the presence of glucose. Furthermore, [18F]F-GAZ is unlikely to be effective as a hypoxia imaging agent. The low in vivo toxicity and substantial retention in tumor observed at high doses of F-GAZ do provide rationale for further testing as a radiosensitizer for external beam radiation therapy of radioresistant, hypoxic tumors.
Introduction
Positron emission tomography (PET), used for diagnosis, staging, and therapeutic monitoring of tumors, 1 has applications in imaging hypoxia, a principal element of the microenvironment of solid tumor. 2 Hypoxia develops from acute or chronic ischemia. It induces a number of vascular, lymphatic, and membrane changes that impair the delivery of oxygen and nutrients to the hypoxic area, 3 –5 and gives rise to a broad range of transcriptional and post-translational cellular adaptations that promote metastases and compromise chemotherapy. 6 –8 A negative association of hypoxia with clinical outcomes strongly supports the need to identify tumors with a high hypoxic fraction, so that hypoxia-directed treatments could be implemented and treatments that are oxygen-dependent can be avoided. 9 –11
Azomycin (2-nitroimidazole, 2-NI) derivatives were initially developed as low-toxicity oxygen mimetic radiosensitizers for radiotherapy of hypoxic tumors. Their intracellular bioreductive activation and subsequent adduct formation with tissue macromolecules results in their accumulation only in hypoxic cells. 12 Despite their low accumulation in most normally oxygenated cells, lipophilic azomycins concentrate in lipoidal tissues such as brain, and at pharmacological doses, cause dose-limiting toxicity. 13,14
Depending on the radioisotope used, 2-NIs for imaging tumor hypoxia can be either single-photon emission computed tomography (SPECT) 15 or PET 16 agents. The misonidazole (MISO) derivative [18F]-Fluoromisonidazole ([18F]FMISO), 17 is the most widely used PET agent for hypoxia imaging, whereas iodoarabinofuranosyl-2-nitromidazole ([123I]IAZA) is an effective SPECT agent. 18 Fluoroarabinofuranosyl-2-nitromidazole ([18F]FAZA), 19,20 was developed to improve image contrast by increasing the rate of clearance from blood and from nontarget tissue. [18F]FMISO and [18F]FAZA undergo minimal metabolism in vivo and are now established PET radiopharmaceuticals.
Despite the widespread clinical application of [18F]FMISO and [18F]FAZA, a low image contrast and longer uptake and retention times resulting in later PET imaging times has stimulated research into the generation of hybrid compounds. One idea included the utilization of facilitated transport into hypoxic cells via hypoxia-upregulated glucose transporters (e.g., GLUT1) and possible additional trapping by hexokinases. 21 However, L18F-labeled 2-fluoro-6-deoxy-D-glucose ([18F]FDG) coupled to 2-NI as the C-1 glucoside ([18F]FDG-NI), did not accumulate in a murine hypoxic tumor model in vivo. More recently, a glucose conjugate of diacetyl-bis(N4-methylthiosemicarbazone (64Cu-ATSM-G), also a C1 glucoside, was reported to retain the hypoxia-selective profile of 64Cu-ATSM in vitro, but in vivo, in comparisons to [18F]FDG and 64Cu-ATSM, it displayed diffusion-like uptake patterns similar to 64Cu-ATSM. It has also been reported that methyl glucoside and other fluoroalkylated glucosides are not GLUT1 substrates. 22,23
In contrast to glucose derivatization at C1, C6 derivatives that are not glucosides have been studied. 6-Fluoro-6-deoxy-D-glucose (6-[18F]FDG) 24,25 and 6-iodo-6-deoxy-D-glucose (6-IDG) are reported to be GLUT1 transport substrates 26 , but when an iodophenyl group was introduced at C6-O-, the glucose transporter no longer recognized it. 27 On the other hand, C6 nitrobenzoxydiazoamino glucose (6-NBDG) retains some interaction with GLUT1, but its transport capability is greatly reduced compared to D-glucose. 28
A new glucosyl-azomycin adduct in which 2-NI is coupled to glucose at C6-O- via a flexible aliphatic linker has been reported. 29 By analogy to 6-NBDG, this compound would be a putative substrate for GLUT1 transport. The radiosynthesis of N-(2-[18F]fluoro-3-(6-O-glucosyl)propyl)-2-nitroimidazole ([18F]fluoroglucoazomycin; [18F]F-GAZ; Fig. 1), and its in vitro and in vivo uptake and biodistribution in the murine EMT-6 breast tumor model, are now described.
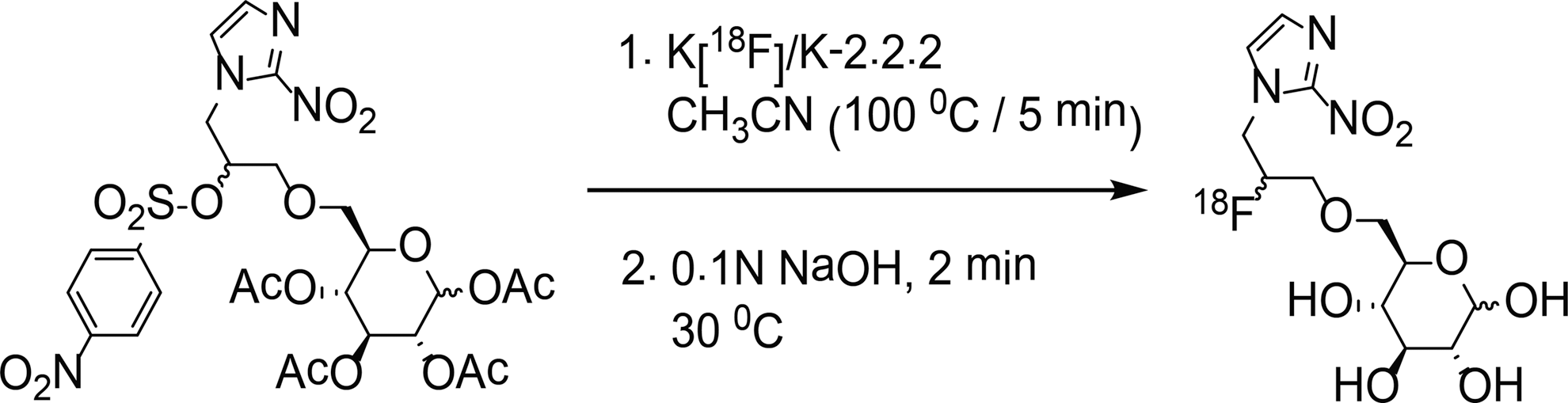
Radiosynthesis of [18F]F-GAZ. [18F]F-GAZ, N-(2-[18F]fluoro-3-(6-O-glucosyl)propyl-azomycin.
Materials and Methods
All standard chemicals used were purchased from Fisher Scientific or Sigma Aldrich.
[18F]F-GAZ radiosynthesis
The radiosynthesis of (3R,4S,5S,6R)-6-((2-[18F]-fluoro-3-(2-nitro-1H-imidazol-1-yl)propoxy)methyl)tetrahydro-2H-pyran-2,3,4,5-tetraol; [18F]F-GAZ) was carried out starting from its tetra-acetyl nosylated precursor 29 using an automated synthesis unit (ASU; GE Tracerlab™). The nosyl precursor (∼5 mg) was dissolved in anhydrous acetonitrile (1 mL) and reacted with the anhydrous potassium [18F]fluoride–Kryptofix-2.2.2 complex for 5 minutes at 100°C, followed by base-catalyzed (0.1 N NaOH) deacetylation of the labeled precursor (Fig. 1). [18F]F-GAZ was recovered from the reaction mixture by high-performance liquid chromatography (HPLC) radiochromatography (Luna II, 10 μ 100 A, 25×0.9-cm C-18 reverse-phase column; mobile phase 8% ethanol in water; flow rate 1.5 mL/min), at a retention time of ∼23 minutes. Quality control of the HPLC-purified product by thin layer radiochromatography (TLC) on silica gel microTLC analytical plates (2.5×7.5 cm; Whatman) developed with H2O:ethanol (9.25:0.75, v/v) and radiometrically scanned using a Model AR2000 plate scanner (Bioscan) revealed a single radioactive species at Rf=0.40, as for the unlabeled reference material.
Animals and toxicity
All animal experiments were carried out in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the local animal care committee (Cross Cancer Institute, Alberta Health Services, Edmonton).
The overt toxicity of F-GAZ in BALB/c mice was investigated before undertaking the high-dose imaging experiment. In a longitudinal dose escalation design, mice (n=3) initially received single intraperitoneal (i.p.) doses of F-GAZ (50 mg/kg). Animals were observed for 24 hours for any indications of toxicity. In the absence of visible signs of toxicity, this dose/24 hours observation protocol was repeated sequentially at escalating doses of 300, 600, and 900 mg/kg (maximum tolerated dose [MTD]), using three new mice at each dose level. All mice tested were then observed for an additional 2 weeks after dosing. Heart, liver, kidney, and brain were collected immediately upon CO2 euthanization for gross and histopathological examination by a certified veterinary pathologist. No pathologies attributable to the drug toxicity were observed in any of the 12 mice.
For in vivo tumor model studies, murine EMT-6 cells (2–5×106 cells in 100 μL phosphate-buffered saline) were injected subcutaneously into the upper left flank of female BALB/c mice (20–24 g; Charles River). The EMT-6 tumor-bearing mice were used for PET imaging and biodistribution experiments after allowing 8 to 11 days for the tumors to reach sizes of about 250 mm3.
Small animal PET-imaging studies
PET experiments were performed on EMT-6 tumor-bearing BALB/c mice fasted for 3–4 hours before the imaging experiments. The animals were anesthetized (inhalation of isoflurane in 40% oxygen/60% nitrogen; 1 L/min) and the body temperature was maintained at 37°C. Mice were immobilized in the prone position, with their medial axis parallel to the axial axis of the scanner and their thorax, abdomen, and hind legs (organs of interest: heart, kidneys, bladder, and tumors) in the center of the field of view of the Concorde microPET® R4 scanner (Siemens Preclinical Solutions). A transmission scan for attenuation correction was not acquired. The amount of radioactivity present in the injection solution (nominally 7.5 MBq;≈0.04 ng) in a 0.5-mL syringe was determined with a dose calibrator (Atomlab™ 300, Biodex Medical Systems), which was cross calibrated with the scanner. The 3D list mode emission scan (60 minutes acquisition) was started, and after a delay of approximately 15 seconds, [18F]F-GAZ (3–5 MBq in 100–150 μL saline) was injected through a needle catheter into the tail vein. For low-specific-activity studies, unlabeled F-GAZ dissolved in saline (final concentration of 2 mg per 20 g, corresponding to a dose of 100 mg/kg) was coinjected with the radiotracer [18F]F-GAZ.
The list mode data were sorted into sinograms (53 time frames; 10×2 seconds, 8×5 seconds, 6×10 seconds, 6×20 seconds, 8×60 seconds, 10×120 seconds, and 6×300 seconds). The frames were reconstructed using the Ordered Subset Expectation Maximization applied to the 2D sinograms (2D OSEM). The pixel size was 0.085×0.085×0.12 cm, and the resolution in the center field of view was 1.8 mm. No correction for partial volume effects was performed. The image files were further processed using the ROVER v2.0.30 software (ABX GmbH). Masks for defining 3D regions of interest (ROI) were set and the ROIs were defined by thresholding. ROI time–activity curves (TACs) were generated for the subsequent data analysis. Standardized uptake values (SUV=(activity/mL tissue)/(injected activity/body weight), mL/g) were calculated for each ROI.
Biodistribution experiments
Biodistribution studies were performed in EMT-6 tumor-bearing BALB/c mice after finishing the PET experiment at 60 minutes post-injection (p.i.), using the high-specific activity (no carrier added) and the low-specific activity (carrier added) [18F]F-GAZ. At 60 minutes p.i., the animals were euthanized by cervical dislocation and rapidly dissected. Organs of interest included blood, heart, lung, liver, kidneys, gall bladder, spleen, duodenum, small and large intestine, pancreas, right femur, muscle, ovaries, brain, fat and tumors, which were collected and weighed. Radioactivity in all tissues was measured in a γ-counter (Wallac 1480 Wizard-3, Perkin-Elmer), and results were analyzed as percentage of injected dose per gram of tissue (%ID/g).
In vitrol cell experiments
EMT-6 cells were cultured at 37°C in a humidified atmosphere of 5% (v/v) CO2, using the DMEM/F12 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 1% antibiotic/antimycotic (Invitrogen). The cell growth medium was changed every second day and cells were tested against mycoplasma contamination. Exponentially growing cells were subcultured into 12-well plates (Corning) 24 hours before the radiotracer uptake experiment, at a density that gave rise to >85% confluence on the cell implantation day. Before implantation, the growth medium was removed, and the cells were washed once with the Krebs-Ringer buffer (120 mM NaCl, 25 mM NaHCO3, 4 mM KCl, 1.2 mM KH2PO4, 2.5 mM MgSO4, and 70 μM CaCl2, pH 7.4). The cells were subsequently incubated in the Krebs-Ringer buffer for 1 hour at 37°C before radiotracer incubation was started. To estimate the affinity of F-GAZ for GLUT1 in comparison to that of D-glucose and 2-FDG, half-maximum inhibition coefficients (IC50) of the three compounds were determined. The EMT-6 cells were incubated with either the glucose-free Krebs-Ringer buffer containing [18F]FDG and various concentrations F-GAZ (10−5–10−1 M) or increasing concentrations of D-glucose or 2-FDG (10−7–3×10−2 M) or no compound at all for comparison (=100% uptake of [18F]FDG). After 60 minutes, cells were rinsed with ice-cold Krebs-Ringer solution, lysed, and counted in a γ-counter. Cell uptake levels were normalized to percent of the total uptake of [18F]FDG without any compound present (control).
Data analysis
All data are expressed as means±SEM. TACs were constructed using GraphPad Prism® 4.0 (GraphPad Software). Where applicable, statistical differences were tested by the Student's t-test and were considered significant for p<0.05. Molar concentrations producing half-maximum inhibition of the maximum uptake (IC50) of F-GAZ, D-glucose, and 2-FDG in EMT-6 cells were determined by nonlinear regression.
Results
Radiochemistry
The synthesis of F-GAZ and its tetra-acetylated GAZ nosylate precursor were described previously. 29,30 Radiofluorination of the tetraacetyl GAZ nosylate precursor was performed according to the scheme in Figure 1 using an ASU, followed by HPLC purification. In a typical reaction, [18F]fluoride afforded crude [18F]F-GAZ in 34 minutes. [18F]F-GAZ was purified by HPLC in 40 minutes, resulting in a total synthesis time of 74 minutes. The radiochemical yield resulted in <1%, which was sufficient for radiopharmacological experiments. [18F]F-GAZ had a radiochemical purity of≈98% on TLC. The radiochemistry involved the nucleophilic replacement of the nosyl group located at C8 (linker) of the tetraacetyl nosylate precursor. The radiofluorination was conceived from the diastereomeric mixture of the nosylate, therefore this reaction afforded a mixture of 1-α/β-D-(6-I-(9-[2-nitro-1H-imidazolyl]-8S-[18F]fluoropropyl)glucopyranose and 1-α/β-D-(6-O-(9-[2-nitro-1H-imidazolyl]-8R-[18F]fluoropropyl)glucopyranose diastereomers. The nosylate precursor was used as the enantiomeric mixture and [18F]F-GAZ was recovered and used without separation of the isomers.
Dynamic small animal PET imaging
In vivo uptake, biodistribution and clearance parameters of [18F]F-GAZ were evaluated in EMT-6 tumor-bearing mice. Images (Fig. 2) represent time intervals from 15 seconds to 60 minutes p.i. The radioactivity was rapidly cleared from the body, resulting in large amounts in the urinary bladder within 5 minutes. Uptake by the EMT-6 tumor was visible, especially after 15 minutes p.i., but did not increase over time, resulting in only faint delineation of the tumor for all time points after 15 minutes. TACs were generated from the PET image uptake, analyzed as SUV, for the EMT-6 tumor and muscle tissue, and the tumor–muscle ratios were derived (Fig. 3) from the SUVs. The TAC for the tumor revealed fast, but low-level uptake of [18F]F-GAZ, reaching a maximum SUV of 0.66±0.05 (n=6) after 5 to 6 minutes post-injection. The SUV values decreased over the time course of the experiment, to 0.24±0.04 after 60 minutes (n=6; p<0.001). A similar pattern was observed in muscle tissue, although at somewhat lower levels: 0.49±0.04 after 5 minutes and 0.14±0.03 after 60 minutes (n=6; p<0.001). The curves for [18F]F-GAZ were compared to the previously reported TACs for [18F]FAZA to analyze the in vivo uptake and retention of [18F]F-GAZ in this tumor model. 31 In contrast to [18F]F-GAZ, the hypoxia PET tracer [18F]FAZA accumulated slowly, remained at a constant level in the tumor cells for some time, and then began to slowly diffuse out. [18F]F-GAZ muscle clearance was more pronounced than tumor clearance, resulting in a continuous increase in tumor–muscle ratios after 30 to 40 minutes p.i. The TACs for [18F]F-GAZ in the heart (analyzed as blood pool), kidneys, and bladder are depicted in Figure 4. Radioactivity was cleared rapidly from the blood via the kidneys.

Representative dynamic small animal positron emission tomography images (up to 60 minutes post-injection) of [18F]F-GAZ in an EMT-6 tumor-bearing BALB/c mouse after injection of 7.5 MBq.
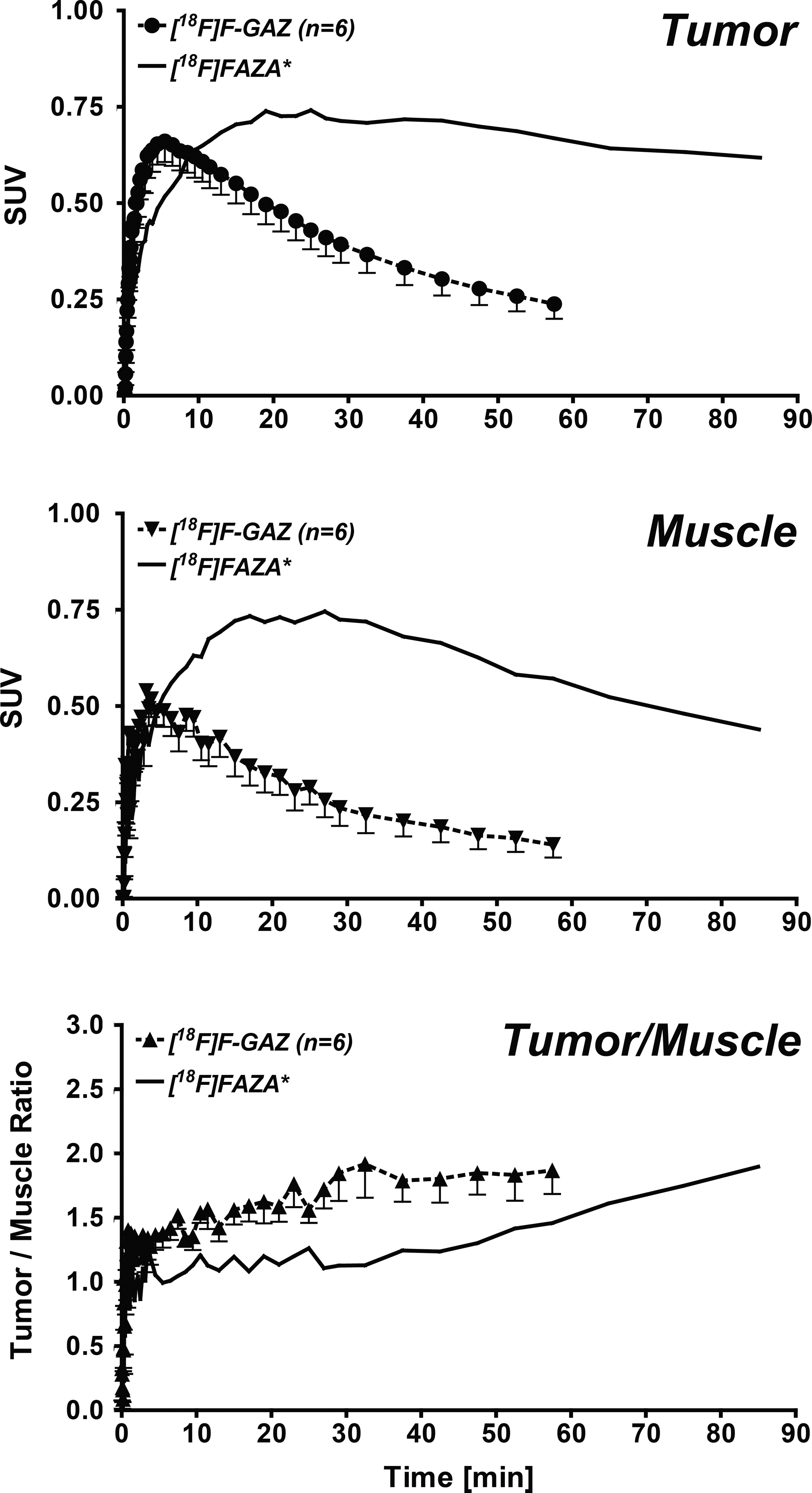
TACs for the radioactivity profile of [18F]F-GAZ in EMT-6 tumors, muscle tissue, and tumor–muscle ratios. Data are shown as SUV (top, middle) or as tumor/muscle ratios (bottom) and as means±SEM from 6 tumor-bearing mice. *For comparative purposes, TACs for [18F]FAZA, analyzed in the same murine tumor model, were included from a different work. 32 TAC, time–activity curves; SUV, standardized uptake values.
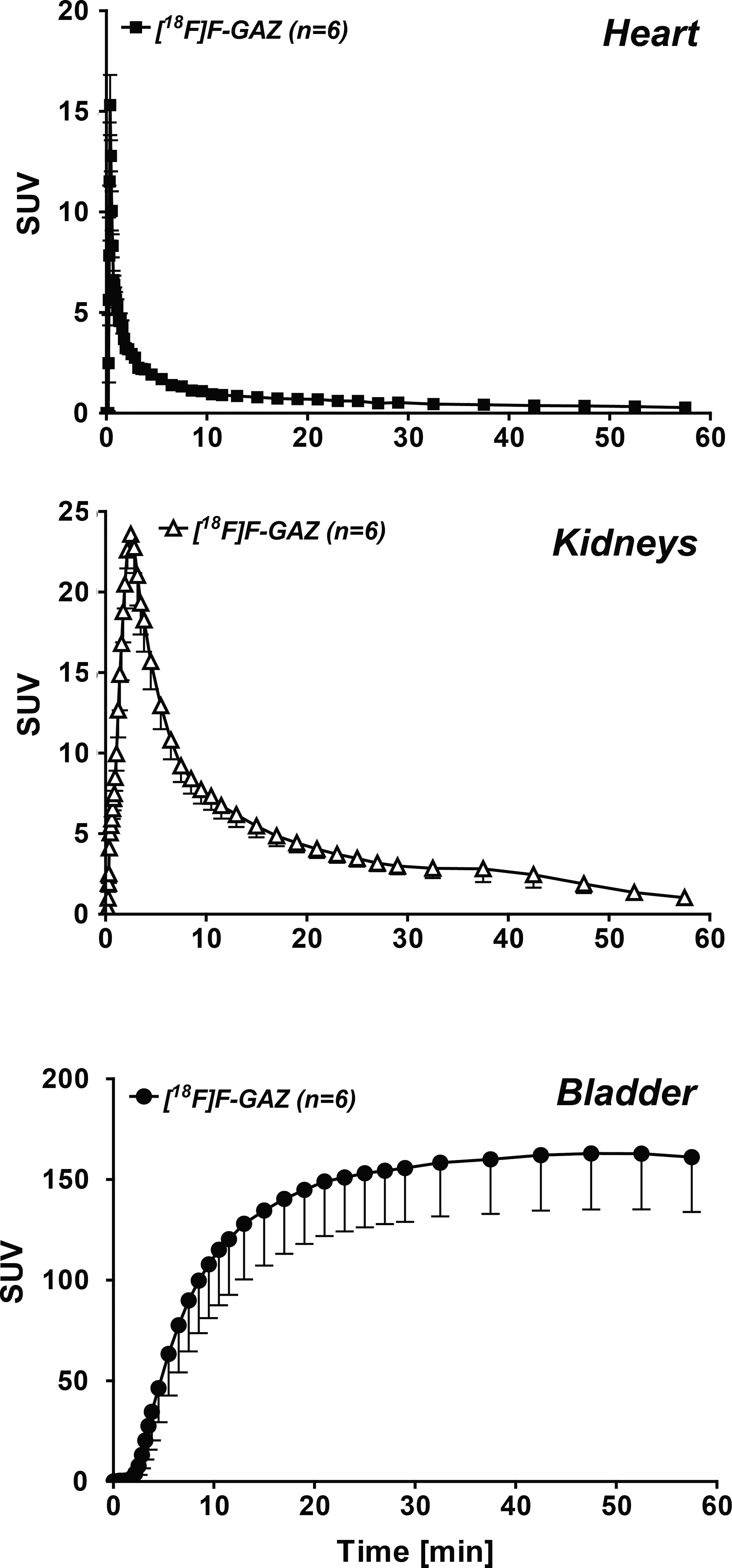
TACs of the radioactivity profile of [18F]F-GAZ in heart (blood pool), kidneys, and bladder. Data are shown as SUV and means±SEM from 6 EMT-6 tumor-bearing BALB/c mice.
Toxicity
Before injection of pharmacological doses of F-GAZ (cold F-GAZ added to [18F]F-GAZ), the in vivo MTD was determined. No evidence and pathological signs of toxicity were found after single i.p. doses ranging from 50 to 900 mg/kg.
Biodistribution of the radiotracer
The biodistribution of [18F]F-GAZ was determined in EMT-6 tumor-bearing mice immediately after the PET experiments at 60 minutes post-injection (Fig. 5). Uptake in liver, kidneys, duodenum, intestines, gall bladder, and pancreas all exceeded 1%ID/g, and tumor uptake was 1.25%±0.15%ID/g (n=4). Images (not shown) acquired after dosing with low-specific activity [18F]F-GAZ (i.e., with carrier F-GAZ; 100 mg/kg) were qualitatively identical to those for high-specific activity [18F]F-GAZ, and except for the kidneys, duodenum, and bone, the biodistribution of low-specific activity [18F]F-GAZ was generally similar to that of high-specific activity [18F]F-GAZ (Fig. 5), with no significant change of the radiotracer's tumor uptake (%ID/g 0.99±0.13; n=3; p>0.05). At a high dose, renal concentrations reached ∼7%ID/g, almost triple the values for the high-specific-activity study, whereas liver, gall bladder, and intestinal tract radioactivity (as_%ID/g) was slightly lower for the low-dose group.
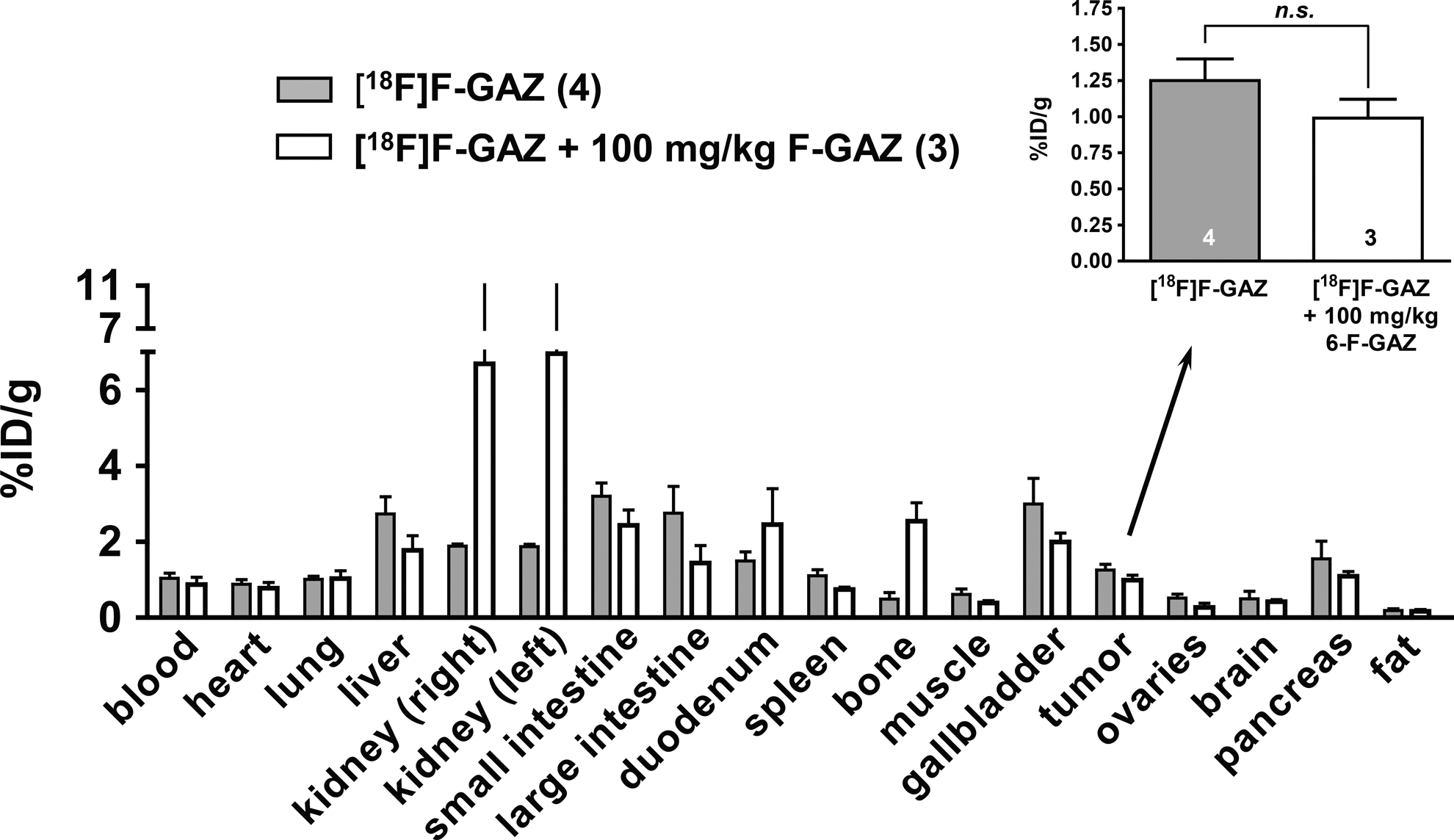
Biodistribution of [18F]F-GAZ alone and in the presence of 100 mg/kg unlabeled F-GAZ after 60 minutes post-injection. Data are shown as means±SEM from EMT-6 tumor-bearing BALB/c mice. The inset shows the biodistribution of 6-[18F]F-GAZ in the EMT-6 tumors only; in the absence (n=4) and in the presence (n=3) of 100 mg/kg unlabeled F-GAZ in the mice.
Effects on [18F]FDG uptake in vitro
In vitro experiments in EMT-6 cells were performed using unlabeled F-GAZ to test the initial hypothesis that F-GAZ would be a substrate for facilitative glucose transporters (e.g., GLUT1). For this purpose, cell uptake of [18F]FDG, known to occur mainly through GLUT1, 32 was studied. Inhibition of [18F]FDG uptake by increasing concentrations of D-glucose, 2-FDG, and F-GAZ were analyzed and compared. As expected, D-glucose inhibited cell uptake of [18F]FDG in a concentration-dependent manner, with an IC50 of 210±20 μM (n=9/3, Fig. 6). At concentrations of 1 mM and higher, unlabeled F-GAZ also competitively blocked uptake of [18F]FDG, with an IC50 of 37±4 mM (n=6/2), a two order of magnitude higher concentration range. For comparison, the IC50 for unlabeled FDG (2-FDG) was found to be 181±17 μM (n=6/2), similar to that for D-glucose.

Concentration–response curves for F-GAZ, D-glucose, and 2-FDG for their inhibiting effects on the cell uptake of [18F]FDG into EMT-6 cells. Data are shown as_% maximum radiotracer uptake (control=100%) and as means±SEM from n/x different experiments (n being number of investigated wells, x being number of experimental days).
Discussion
Radiosensitizing azomycins enter cells via passive diffusion and are metabolically trapped in cancer cells only under hypoxic conditions. Their overall biodistribution patterns and accretion into tumor are influenced by lipophilicity, rates of renal and hepatic clearance from blood, metabolic biotransformation, and protein binding.
Novel, hydrophilic, bioreductively activated compounds, which would be cleared rapidly from blood and nontarget tissues, and which would undergo active or facilitative transport into hypoxic cells via glucose 21,33,34 and peptide 35 transporter proteins, have been reported. Of these, F-GAZ, a hydrophilic, oxygen-mimetic hypoxic cell radiosensitizer 29 was designed to exploit hypoxia-upregulated facilitative glucose transporters (GLUTs), 36 to attain higher target-tissue concentrations than realized with the current generation of hypoxia-selective nitroimidazoles.
The data reported here for [18F]F-GAZ represent average results for the four diastereomers generated during the radiofluorination step. It has been reported that there are only small differences among these isomers both in their modeled GLUT-1-binding energies and in their modeled bioreductive properties (E LUMO). 30 Therefore, no separation was attempted before use in the current proof of principle study.
MISO, a promising radiosensitizer, was clinically ineffective due to dose-limiting toxicities at doses of 10–12 g/m2. 37,38 Less lipophilic nitroimidazoles like SR2508 were less toxic, but also less efficacious at tolerated doses. 39 In comparison, mice receiving F-GAZ in single i.p. doses of up to 900 mg/kg showed no evidence of acute toxicity. Studies with carrier-added [18F]F-GAZ (0.286 mmol/kg; 100 mg/kg) produced tumor concentrations equivalent to∼1% ID/g 1 hour after i.v. injection. This amounts to∼200 μg (0.57 μmol) per g tumor, an in vivo concentration akin to the F-GAZ concentration (0.5 mM) that produced a sensitizer enhancement ratio of ∼1.2 for EMT-6 cells in vitro. 29 Thus, it appears that in vivo radiosensitizing concentrations of F-GAZ are attainable through i.v. bolus or infusion dosing.
[18F]F-GAZ was ubiquitously distributed throughout the body and rapidly cleared via the kidneys in Balb/c mice during the first hour after injection. Rapid egress of radioactivity from tumor, despite retaining ∼1%ID/g at 60 minutes after injection, produced low-contrast PET images, unlike the classical radioactivity retention profiles following [18F]FAZA injection in the same tumor model (Fig. 3). This indicates that little or no hypoxic trapping of [18F]F-GAZ occurs in this hypoxic tumor model, compared to the uptake and retention of [18F]FAZA. 31 On the other hand, some uptake of [18F]F-GAZ occurred, with a maximum detected SUV of 0.66 for [18F]F-GAZ (5 minutes p.i.) versus 0.74 for [18F]FAZA. The faster [18F]F-GAZ tumor uptake kinetics in comparison to [18F]FAZA indicate that the uptake mechanism of [18F]F-GAZ must be different from the passive diffusion mechanism of [18F]FAZA. 31 [18F]F-GAZ radioactivity was retained after 60 minutes p.i., with tumor–muscle ratios still higher than 1.
The biodistribution, excretion, and imaging patterns of [18F]F-GAZ in the EMT-6 Balb/c model were relatively independent of dose, ranging from subpicogram to 100 mg/kg (0.59 pmol/kg to 286 μmol/kg; Fig. 5). Other oxygen mimetic radiosensitizers, for example, [123I]IAZA (0.18–18 mg/kg; 0.5–50 μmol/kg) 15 and [131I]IAZGP (0.002–2 μmol/kg), 40 display dose-independent uptake into hypoxic tumors. This means that the PET-imaging data for tracer doses of these oxygen mimetic radiosensitizers could be used to model pharmacokinetics for the therapeutic doses used in XRT/radiosensitization protocols and for magnetic resonance imaging (100–200 μmol/kg) of fluorinated radiosensitizers. 41 The dose-independent biodistribution of [18F]F-GAZ also implies that the absorption, bioreduction, and adduct-formation mechanisms are not saturable, at least not at the maximum dose tested (100 mg/kg; 286 μmol/kg). This would leave diffusion, rather than facilitated transport, as the main uptake mechanism for F-GAZ, in agreement with the in vitro data, which show that F-GAZ is a weak competitor of [18F]FDG uptake by EMT-6 cells.
A review of structure–activity relationships between substituted glucoses and glucose, transport proteins (GLUTs) lies well beyond the scope of this work. It is noteworthy, however, that glucose molecules with 123I-containing substituents or organometallic technetium and rhenium complexes were not transported by GLUT1, 26,27,42 and that glucosides (C1-glucose conjugates) are generally considered substrates of active (sodium-dependent) transporters (i.e., SGLTs) rather than facilitative transporters (GLUTs). 23 Reported glucose-conjugated radiosensitizer/hypoxia-imaging agents include [18F]FDG-NI, 21 64Cu-ATSM glucoside, 33 and several members of a family of azomycin glycosides tethered via a 5 member amide-containing linker. 32 Tumor uptake of [18F]FDG-NI by a rat mammary carcinoma model was low, with rapid clearance from tumor tissue within 60 minutes, with a tumor–blood ratio (T/B) of 1 and a tumor–muscle ratio (T/M) of 2.3. This is similar to the data now reported for [18F]F-GAZ, in the EMT-6 model, with T/B=1.2 and T/M=2.0, all determined 60 minutes p.i. Comparison of the in vivo properties of both compounds indicates that substitution of C1 with azomycin alone, or C6-O- with a longer aliphatic linker plus azomycin, leads to similar tumor uptake and clearance patterns in vivo. Although [18F]F-GAZ is putatively a GLUT substrate and [18F]FDG-NI is a putative SGLT substrate, they exhibit similar in vivo biodistribution and clearance patterns, implying that their mechanisms of uptake into tissue may be based on diffusion rather than facilitative or active transport. FDG-NI in D-glucose-free buffer in vitro inhibited uptake of [18F]FDG by Walker 256 tumor cells at concentrations of 0.5 mM and higher, 21 a concentration range similar to that now reported for the inhibition of [18F]FDG uptake by F-GAZ in EMT-6 cells (IC50 of 37±4 mM), but at concentrations two orders of magnitude higher than D-glucose and 2-FDG. This suggests that both hybrid compounds interact weakly with one or more transporters of FDG, including GLUT1, which is the main GLUT subtype responsible for uptake of [18F]FDG into EMT-6 cells. 43 FDG is also transported, albeit weakly, by SGLTs, 44 but expression of SGLTs in EMT-6 cells is unknown. In any case, at microdoses used for PET radiotracers, neither F-GAZ nor FDG-NI can compete with physiological concentrations of D-glucose in vivo, and therefore their uptake via glucose transporters into tumor cells is unlikely to occur.
Mechanisms of uptake of F-GAZ may be analogous to the modeled relationship between GLUT1 and 6-NBDG, 28 a glucosyl-6-O-adduct with some structural similarities to F-GAZ. The estimated Km of 0.26 mM of 6-NBDG was based on experiments done in GLUT1-rich astrocytes. Compared to the Km for D-glucose (5 mM) for GLUT1 as determined in expression systems, 6-NBDG would appear to have a higher affinity to GLUT1 than D-glucose itself, although its uptake and transport into astrocytes was very slow even in the absence of D-glucose. 28 Indeed, preliminary affinity modeling data indicate that 6-NBDG and F-GAZ have similar binding energies for GLUT1 (−8.35 kcal/mol and −7.79 kcal/mol, respectively), and which are substantially higher than the −5.30 kcal/mol calculated for glucose in the same model. 30 These modeling data provide further support that F-GAZ, despite its apparent high affinity for the transporter protein, may, like 6-NBDG, bind strongly, but still be only poorly transported by GLUT1. The current [18F]F-GAZ data and [18F]FDG-NI literature data are derived from whole-cell systems in which the expression of glucose transporters has not been mapped, and therefore preclude more specific statements about the involvement of GLUT1 or SGLTs.
In summary, [18F]F-GAZ is characterized by rapid, ubiquitous distribution in mice following i.v. administration. Rapid renal clearance was evident within the first minute. Concentrations of radioactivity in other organs and the EMT-6 tumor reflected classical diffusion–perfusion profiles, with fast uptake followed by more gradual washout. F-GAZ was not toxic to mice at high (900 mg/kg) i.p. doses. Although rapidly cleared from blood, a single i.v. dose of F-GAZ (100 mg/kg) was able to provide concentrations in tumor at which radiosensitization can be expected to occur (0.5 mM). F-GAZ was a weak competitor of [18F]FDG uptake in cell culture.
Conclusions
F-GAZ was not toxic to mice at high (900 mg/kg) i.p. doses. Concentrations in tumor following pharmacological doses of F-GAZ support further investigation of its radiosensitizing potential in vivo. In contrast, a microdose, typically used for PET radiotracers, failed to provide evidence of hypoxia-based retention of [18F]FGAZ in tumors. In vitro competition studies against [18F]FDG appear to rule out an effective facilitative or active transport role in the cellular uptake of F-GAZ in an in vivo system.
Footnotes
Acknowledgments
The authors would like to thank Dr. John Wilson, David Clendening, and Jayden Sader from the Edmonton PET Center for providing 18F, Ali Akbari for assistance using automated synthesis units, Angela Westover from the Edmonton Radiopharmaceutical Center (ERC) for providing unlabeled 2-FDG, Dr. David Murray for providing a stock of EMT-6 cells, Dan McGinn and Gail Hipperson from the Vivarium of the Cross Cancer Institute for helping in the animal handling, and Dr. Nick Nation for toxicological and pathological examination of mice. This work was supported in part by a Proof of Principle grant from the Canadian Institutes of Health Research (CIHR) and a bridging/pilot grant from the Alberta Cancer Foundation (ACF).
Disclosure Statement
No competing financial interests exist.