
Abstract
Radiation is a primary modality in cancer treatment. Radiation can also reduce tumor growth outside the treatment field, often referred to as the abscopal effect. The mechanisms and therapeutic potential of the abscopal effect have not been fully elucidated. We evaluated the role of vaccination directed against a tumor-associated antigen (TAA) in the induction and amplification of radiation induced abscopal effects. Active-specific immunotherapy with a TAA-specific vaccine regimen was used to induce and potentiate T-cell responses against carcinoembryonic antigen (CEA) in combination with local irradiation of subcutaneous tumors. We examined the potential synergy of a poxvirus-based CEA vaccine regimen in CEA-transgenic (Tg) mice in combination with either external beam radiation or brachytherapy of local tumors. The induction of CD8+ T cells specific for multiple TAAs not encoded by the vaccine was observed after the combination therapy. In two tumor models, the antigen cascade responses induced by vaccine and local irradiation mediated the regression of antigen negative metastases at distal subcutaneous or pulmonary sites. Clinically, local control of the primary tumor is necessary and can sometimes prevent metastases; however, irradiation generally fails to control preexisting metastases. These studies suggest that by coupling tumor irradiation with immunotherapy, the abscopal effect can transcend from anecdotal observation to a defined mechanism that can be exploited for the treatment of systemic disease.
Introduction
Radiation is the standard treatment for many cancer types, traditionally used to locally eradicate tumor cells or alter tumor and/or tumor stroma architecture with either curative or palliative intent. Although the expectation that the effects of radiation exposure would be restricted to the cells directly in the treatment field was challenged 20 years ago by observations of radiation-mediated damage to unirradiated cells, the biologic parameters characterizing these bystander responses have yet to be clearly defined. 1,2 One of the defined classes of these bystander responses is the “abscopal effect,” in which radiation treatment of a tumor propagates to tumors outside the irradiated volume. 3
Many foundational biologic events have been hypothesized for the abscopal effect mechanism of action, including (a) the possible role of the immune system, (b) inflammatory mediators and dendritic cell activation, and now (c) induction of the antigen cascade. Antigen cascade had been described as an immunologic response demonstrating epitopes distinct from and non–cross-reactive with the inducing epitope. Our hypothesis was that the antigen cascade was in part responsible for the abscopal effect mediating regression of antigen variant tumors, tumors at distal sites, and micrometastatic deposits.
We evaluated the potential role of active vaccination in the induction and amplification of radiation-induced abscopal effects. Active-specific immunotherapy with a tumor-associated antigen (TAA)–specific vaccine regimen was used to induce and potentiate T-cell responses against carcinoembryonic antigen (CEA) in combination with local irradiation of subcutaneous tumors. Transgenic (Tg) mice expressing the human CEA gene 4 as “self” in tissues similar to expression in humans were used as a more relevant preclinical model. We examined the potential synergy of a poxvirus-based CEA vaccine regimen in combination with either external-beam radiation or brachytherapy of local subcutaneous CEA-expressing tumors.
In the results reported here, we have examined (a) the effect of sublethal doses of local tumor radiation on the expression of the death receptor Fas in vivo, (b) enhanced susceptibility of irradiated tumor cells to vaccine induced cytotoxic T lymphocytes (CTL)–mediated killing, (c) vaccine and radiation induction of T-cell responses to multiple endogenous tumor antigens not encoded by the vaccine (antigen cascade), and (d) the functional consequence of antigen cascade in mediating abscopal tumor regression of an antigen disparate tumor in two different vaccine/tumor models.
Overall, these results revealed that the phenotypic modulation of tumor cells via radiation therapy significantly improved the therapeutic efficacy of a recombinant anticancer vaccine regimen. Localized radiation of subcutaneous tumors in combination with vaccine led to a dramatic influx of CD8+ T cells to the tumor microenvironment and subsequent inhibition of tumor growth. In addition, we demonstrated that the induction of CD8+ T cells specific for multiple TAAs not encoded by the vaccine were observed after the combination therapy. Finally, we showed that the polyclonal T-cell response induced by vaccine and local primary tumor irradiation could functionally mediate the regression of antigen negative metastases at distal subcutaneous or pulmonary sites. Thus, irradiation of tumor cells can facilitate antitumor activity by engaging the lytic capacity of Ag-specific CTL and promote cross-priming of additional T-cell populations; these studies may thus form the rationale for the induction of abscopal effect and subsequent therapy of micrometastatic and/or occult tumors.
Materials and Methods
Tumor cell lines
The murine colon adenocarcinoma cell line, MC38 (H-2b), has been described. 5 MC38 cells expressing human CEA were generated by retroviral transduction with CEA cDNA 5 and are designated MC38-CEA+. LL/2 murine lung adenocarcinoma tumor cells expressing CEA (LL2-CEA) have been described. 6 Cells were maintained in complete medium (DMEM supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 units/mL penicillin, 300 μg/mL G418, and 100 μg/mL streptomycin). Before transplantation to mice, tumor cells were trypsinized, dispersed through a 70-μm cell strainer (Falcon, Becton Dickinson, Franklin Lakes, NJ), and washed twice in Hank's balanced salt solution (HBSS).
Animals
For in vivo studies, 6- to 8-week-old female C57BL/6 mice were used (NCI/Frederick). C57BL/6 mice Tg for human CEA (designated CEA-Tg) were originally obtained from a breeding pair provided by Dr. John Thompson (Institute of Immunobiology, University of Freiburg, Germany). The generation and characterization of the CEA-Tg mouse has been described. 4 Mice were housed and maintained under pathogen-free conditions in microisolator cages.
Tumor irradiation
Mice were injected with 3×105 MC38-CEA+ tumor cells subcutaneously (s.c.) in the right hind leg. Tumors were measured by digital caliper. For radiation treatment, the mice were restrained in a customized jig and placed in a leaded holder such that only the tumor was exposed to the irradiation. The radiation was performed using a Pantax (East Haven, CT) machine at 300 kV, 10 mA, and a dose rate of 157 cGy/min. The dose used (8 Gy) was predetermined to have a minimal effect on the tumors' s.c. growth rate. For brachytherapy, tumors were implanted with a single 125I-brachytherapy seed (Oncoseed, Oncura, Plymouth Meeting, PA). The tumor dose rate for these seeds was 4 cGy/h/seed.
Tumor characterization
Mice were injected with tumor cells s.c. in the quadriceps area of the right hind limb. Nine days after tumor transplant, tumors were irradiated with 8 Gy. In a subset of mice, tumors were excised 72 hours after tumor irradiation, fixed, sectioned at 5 μm, and stained with anti-Fas mAb or an isotype control for immunohistochemical analysis. For microvessel density and T-cell infiltration studies, tumors were frozen, sectioned at 5 mm, and stained with CD31 (PECAM-1) or anti-CD3 mAb. Entire slides were digitally scanned by an Aperio ScanScope CS scanning system (Aperio Technologies Inc., Vista, CA) and analyzed by the Aperio ImageScope Viewer software. The positive pixel count v9 algorithm was used to measure positive tumor regions. Cell-surface staining of Fas from explanted tumors was performed with primary PE-labeled mAb. All mAb were purchased from PharMingen (San Diego, CA). Immunofluorescence was analyzed and compared with the appropriate isotype-matched controls (PharMingen) with a FacScan cytometer using Cellquest software (Becton-Dickinson, Mountain View, CA).
Recombinant poxvirus vaccines
The recombinant vaccinia and fowlpox viruses containing the human CEA gene and the murine B7-1, ICAM-1, and LFA-3 genes (designated rV-CEA/TRICOM and rF-CEA/
Tumor therapy studies
Single site tumors
MC38-CEA+ tumor cells (3×105) were injected s.c. in the quadriceps area of the right hind limb of CEA-Tg mice. Eight days after tumor transplant, mice were vaccinated s.c. once with 1×108 pfu rV-CEA/TRICOM admixed with 1×107 pfu rF-GM-CSF. Tumors in subsets of mice were irradiated with 8 Gy on day 14 after tumor transplant. On day 15, mice were boosted with 1×108 pfu rF-CEA/TRICOM admixed with 1×107 pfu rF-GM-CSF. This booster vaccination regimen was repeated two additional times at 7-day intervals. Tumors were measured daily by digital caliper in two dimensions, and the volumes were calculated as previously described. 9 Animals were sacrificed when any tumor measurement (length or width) exceeded 20 mm.
Multiple site tumors
CEA-Tg mice were transplanted with MC38-CEA+ tumors (3×105, s.c. right flank) on day 0 and MC38 (CEA-) tumors (3×105, s.c. left flank) on day 5. Mice were treated with rV-CEA/TRICOM on day 8 and 8 Gy external beam radiation of the MC38-CEA+ tumor on day 14. Mice were boosted with rF-CEA/TRICOM on days 15, 22, and 29. All vaccines were given with rF-GM-CSF. Tumor volume on each flank was monitored.
Primary and metastatic tumors
CEA-Tg mice were transplanted into two sites with LL2-CEA+ tumors (3×105, s.c. right flank, and 3×105, intravenously [i.v.]) on day 0. Mice were vaccinated with rV-GP70/TRICOM on day 7, boosted with rF-GP70/TRICOM on days 14, 21, and 28. All vaccines were coadministered with rF-GM-CSF. Primary tumors in subsets of mice were implanted with a single 125I-brachytherapy seed on day 13. On day 35, lungs were harvested and pulmonary metastases were enumerated as previously described. 6
Immunologic assays
To evaluate CD8+ T-cell responses, spleens were removed as indicated, dispersed into single-cell suspensions, pooled, and coincubated with either the CEA peptide (10 μg/mL, EAQNTTYL), 10 the H-2Db-restricted peptide p53232-240 (2 μg/mL, KYMCNSSCM), 11,12 or the H-2Kb-restricted peptide p15E604-611 (1 μg/mL, KSPWFTTL, referred as GP70 peptide) 13 for 7 days. Bulk lymphocytes were recovered by centrifugation through a Ficoll-Hypaque gradient. T cells were restimulated with fresh irradiated naive splenocytes and the corresponding peptide for 24 hours. As control peptides, VSVNP (RGYVYQGL) was used for H-2Db-restricted peptides, or ovalbumin257-264 (SIINFEKL) 14 was used for H-2Kb-restricted peptides. Appropriate control peptide responses were subtracted from the specific response. Supernatant fluid was collected and analyzed for murine IFN-γ by cytometric bead array (BD PharMingen) according to the manufacturer's instructions. Detection limit was 4 pg/mL.
Statistical analysis
Where indicated, the results of tests of significance are reported as p values and are derived from the Student t test using a two-tailed distribution. P values were calculated with a 95% confidence interval using the Statview 4.1 (Abacus Concepts Inc, Berkeley, CA) software package. Significant differences in the distribution of flow cytometry analysis data were determined by the Kolmogorov-Smirnov test, using CellQuest software (BD Biosciences). In graphic representations of data, y-axis error bars indicate the standard error. In some cases, the variation is such that the plot symbol obscures the error bars.
Results
The death receptor Fas is upregulated on tumor cells in vivo after radiation treatment
The Fas/Fas ligand (FasL) system has been characterized as an integral process for the maintenance of immune privilege and regulation of immune homeostasis of peripheral lymphoid interactions under both normal and pathologic conditions. 15 –19 We have shown previously that radiation can induce upregulation of Fas expression on tumor cells and enable their destruction by Ag-specific immune effector cells via Fas-dependent mechanisms. 20 Here, we used digital slide scanning to examine if Fas expression on MC38-CEA+ cells could be upregulated by localized, external beam radiation in vivo.
To that end, C57BL/6 mice were injected s.c. with MC38-CEA+ tumor cells. After 7 days, the tumor was radiated (8 Gy) in one subset of mice, while another subset of mice was maintained without radiation. The tumors were excised 72 hours after radiation and prepared for immunohistochemical staining using an anti-Fas mAb (Fig. 1). The irradiated tumor demonstrated intense Fas immunoreactivity compared with the nonirradiated tumor (Fig. 1A). Digital slide scanning determined that Fas expression on cells was increased 2.6-fold after radiation (p=0.03; Fig 1B). The expression of Fas on tumor cells 72 hours after radiation was above 98%.
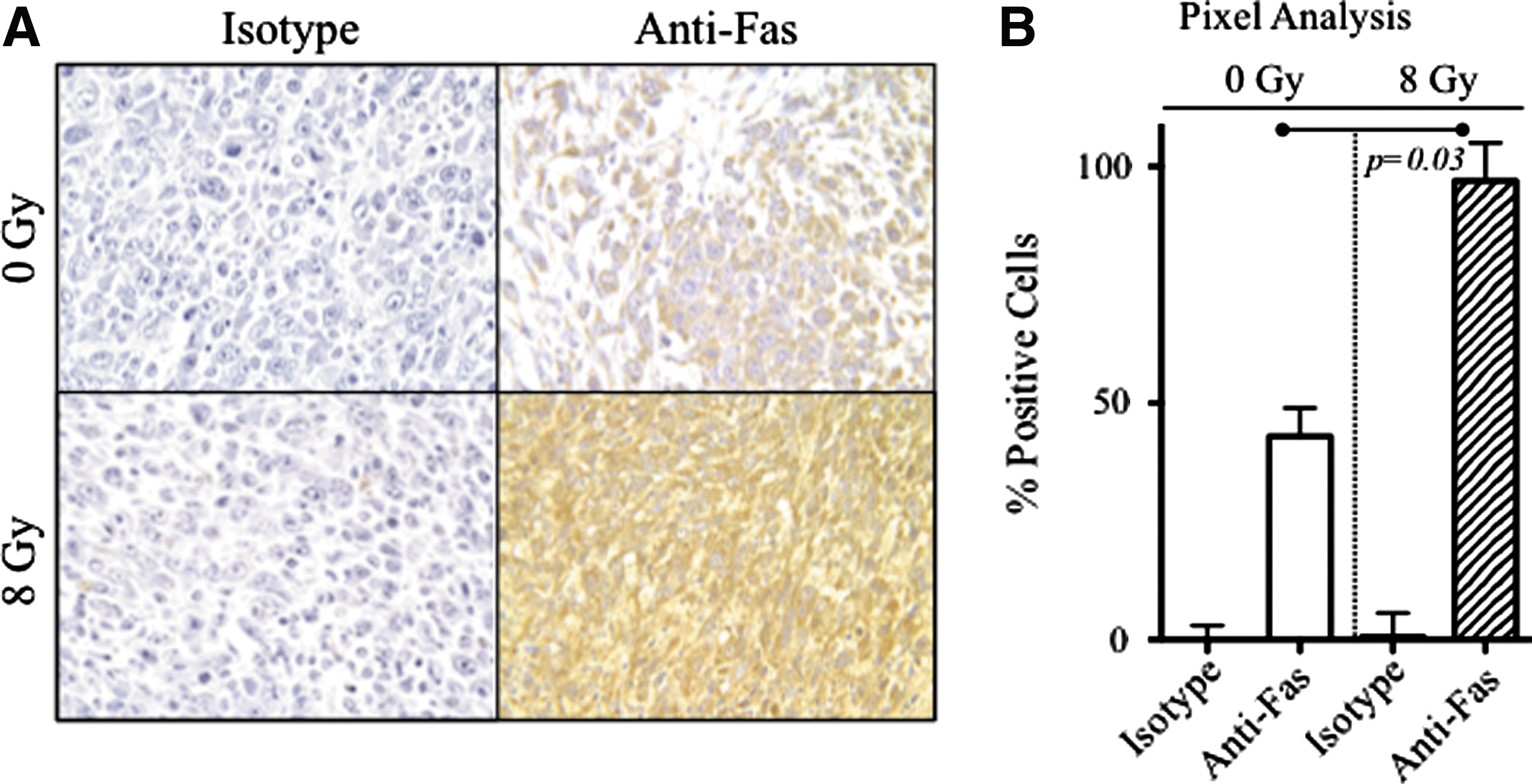
The death receptor Fas is upregulated on tumor cells in vivo after radiation treatment. C57BL/6 mice were transplanted with MC38-CEA+ tumor cells subcutaneously. After 7 days, tumors in a subset of mice were subjected to external beam radiation (8 Gy). Tumors were surgically removed 72 hours after radiation treatment, fixed, sectioned at 5 μm, and immunostained with anti-Fas mAb or an isotype control antibody
Radiation therapy of tumor cells and vaccine therapy resulted in synergistic antitumor activity
To demonstrate whether sublethal radiation of growing tumors improves tumor rejection by a recombinant anticancer vaccine regimen, we vaccinated mice with a diversified prime and boost regimen with CEA/TRICOM vectors in combination with local radiation of the tumor (Fig. 2A). The vaccine regimen consisted of priming mice with rV-CEA/TRICOM admixed with rF-GM-CSF followed by three weekly boosts with rF-CEA/TRICOM admixed with rF-GM-CSF. MC38-CEA+ tumor cells were injected s.c. on the right hind leg of CEA-Tg mice. Eight days after tumor transplant, groups of mice were then divided into those that received (a) no treatment; (b) vaccine alone; (c) irradiation of the tumor alone; or (d) the combination of vaccination followed by radiation.
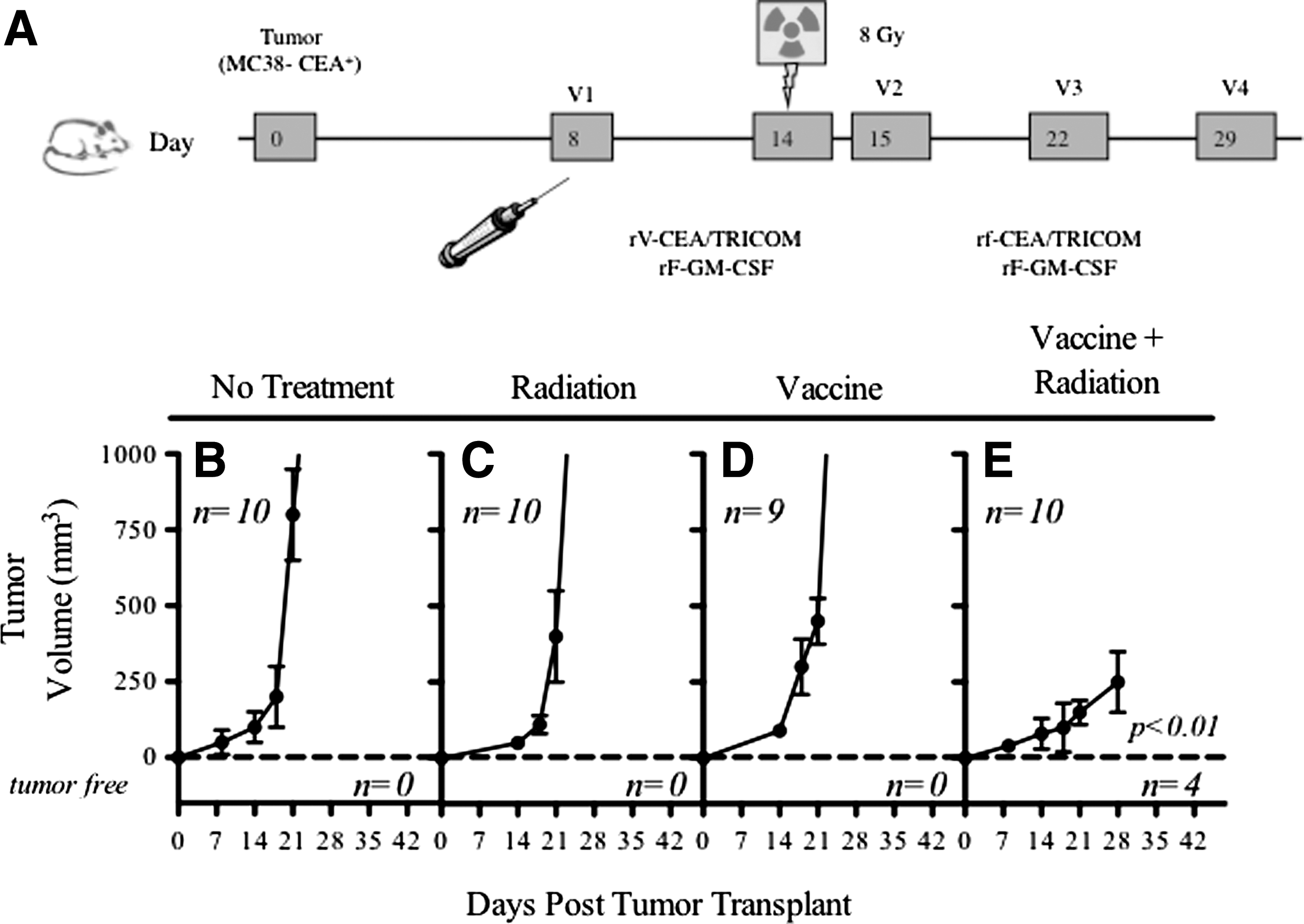
Radiation therapy of tumor cells and vaccine therapy resulted in synergistic antitumor activity. CEA-Tg mice were transplanted with MC38-CEA+ tumor cells subcutaneously.
Tumors of mice that did not receive any treatment grew progressively, ultimately causing the death of the animals (100% by day 30; Fig. 2B). Therapy of tumors with rV-CEA/TRICOM priming on day 8 post-tumor transplant followed by rF-CEA/TRICOM boosting on days 15, 22, and 29 (Fig. 2D) did not significantly inhibit tumor growth. Irradiation of tumors (8 Gy) on day 14 (Fig. 2C) failed to significantly impact the extent of tumor growth in these mice (p=0.682 compared to no treatment). Therapy of tumors with the combination of the vaccine regimen and radiation, however, resulted in a marked and significant decrease in tumor growth rate, and tumor volume (p=0.007 vs no treatment; p=0.001 vs irradiation alone; p=0.001 vs vaccine alone; Fig. 2E). In addition, 40% of the mice treated with the combination of irradiation and vaccine therapy resolved their tumor mass and remained tumor free for the duration of the experiment (180 days). This experiment was repeated four times with similar results.
Characterization of infiltrating cells in tumors treated with the combination of vaccine therapy and radiation
To evaluate the role of tumor architecture and infiltrating immune cells in the tumor microenvironment during combination therapy of vaccine plus radiation, we determined whether changes in tumor vasculature or infiltrating cells were present in the tumor microenvironment after treatment. MC38-CEA+ cells were injected s.c. on the right hind leg of CEA-Tg mice. Eight days later, groups of mice were then divided into those that received (a) no treatment; (b) vaccine alone; (c) irradiation of the tumor alone; or (d) the combination of vaccination followed by radiation.
Tumors were surgically removed 7 days after radiation and analyzed for CD31+ tumor microvessels (Fig. 3A-D) and infiltrating CD3+ T cells by immunohistochemistry (Fig. 3E-H). MC38-CEA tumors are weakly angiogenic (Fig. 3A). In contrast, tumor from mice after radiation demonstrated a significant increase in vascular density as defined by CD31 immunohistochemistry (Fig. 3B, 3I; p=0.002). Tumors from mice that received vaccine alone had no discernible changes in vessel density (Fig. 3C, 3I). Tumors from mice treated with the combination of vaccine followed by radiation, however, displayed the most remarkable and significant modulation of tumor vessel density (Fig. 3D, 3I; p<0.001 vs all groups).
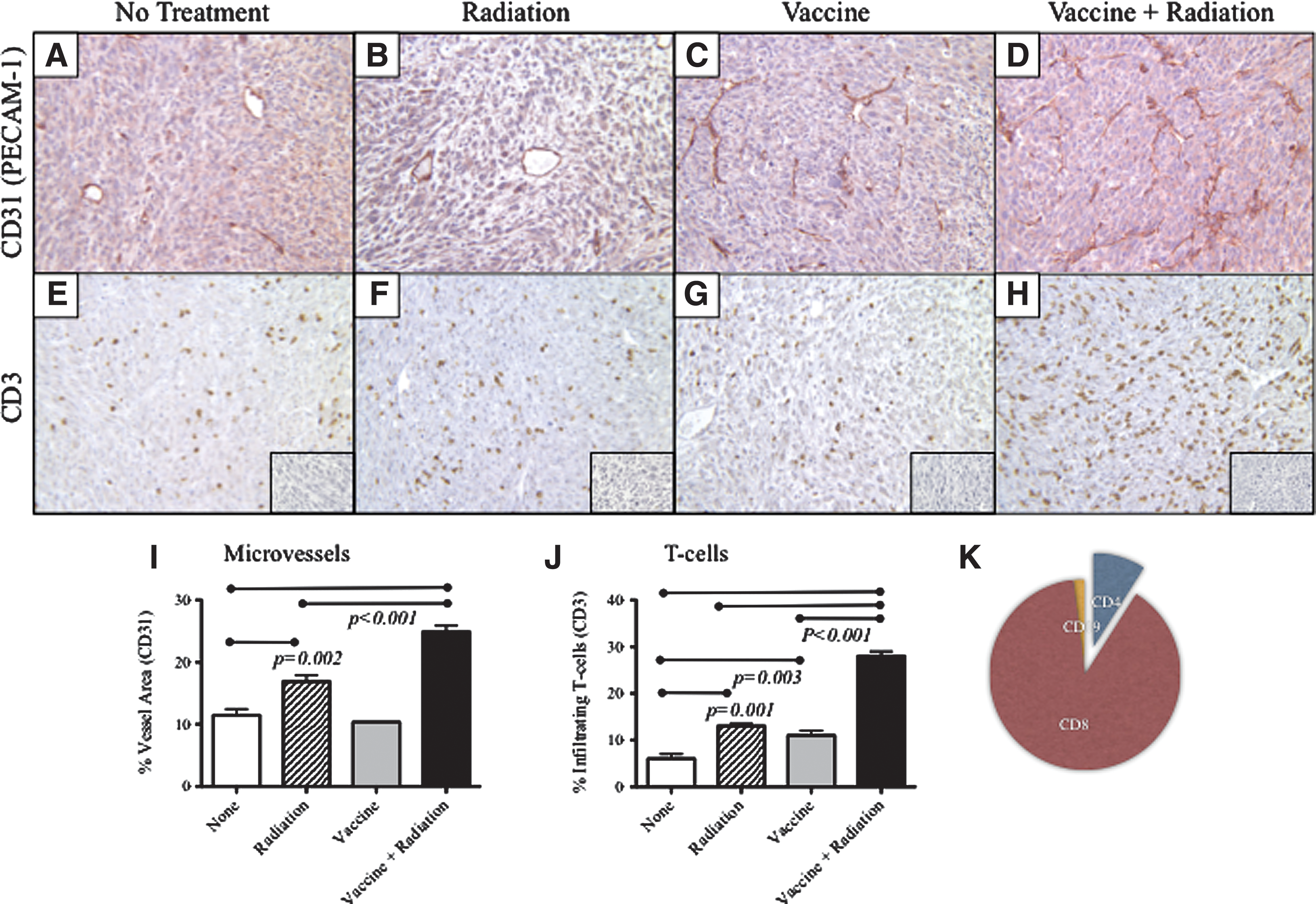
CEA-Tg mice were transplanted with MC38-CEA+ tumor cells. (
Coincident with the marked increase in microvessel density, tumors treated with the combination of the CEA/TRICOM vaccine regimen in combination with radiation demonstrated a substantial increase of T cells in the tumor microenvironment as shown by CD3+ T cells staining by immunohistochemistry (Fig. 3H, 3J). There was a strong correlation between increases in vessel density and T-cell infiltration (R2=0.936). When additional subpopulations of infiltrating cells were quantified, it was observed that CD8+ T cells comprised the majority of the infiltrating CD3+ T cells (93%), with CD4+ T cells (4%) and CD19+ B-cells (3%) comprising the minority. These cell ratios (1:0.04:0.03) remained constant for all groups, regardless of the treatment regimen (Fig. 3K). Collectively, these data show that radiation and subsequent T-cell infiltration transform the weakly angiogenic vasculature into a dense network of functional small vessels of normal appearance.
CD8+ T-cell responses to multiple tumor antigens after therapy with vaccine and radiation
Cellular immune responses were monitored after treatment with the combination of vaccine therapy and radiation. MC38-CEA+ tumor cells, in addition to expressing CEA, have been reported to express the gene products of endogenous murine retroviruses, 21 including GP70, 13,22 which could behave as targets of the CTL response. In addition, these cells have been shown to overexpress wild-type p53, another potential target antigen. 11 Tumor-bearing CEA-Tg mice treated with the combination of the CEA/TRICOM vaccine regimen plus irradiation and cured of established tumor, as described above (Fig. 2E), were monitored for 6 months. At that time, CD8+ T-cell responses in spleen specific for multiple TAAs were measured from the tumor-free mice. Because no mice in the untreated groups survived (Fig. 2A), we compared all responses to normal age-matched CEA-Tg mice. CD8+ T cells from mice treated with the combination of vaccine and irradiation showed significantly higher CEA-specific IFN-γ production compared with the control group (Fig. 4).
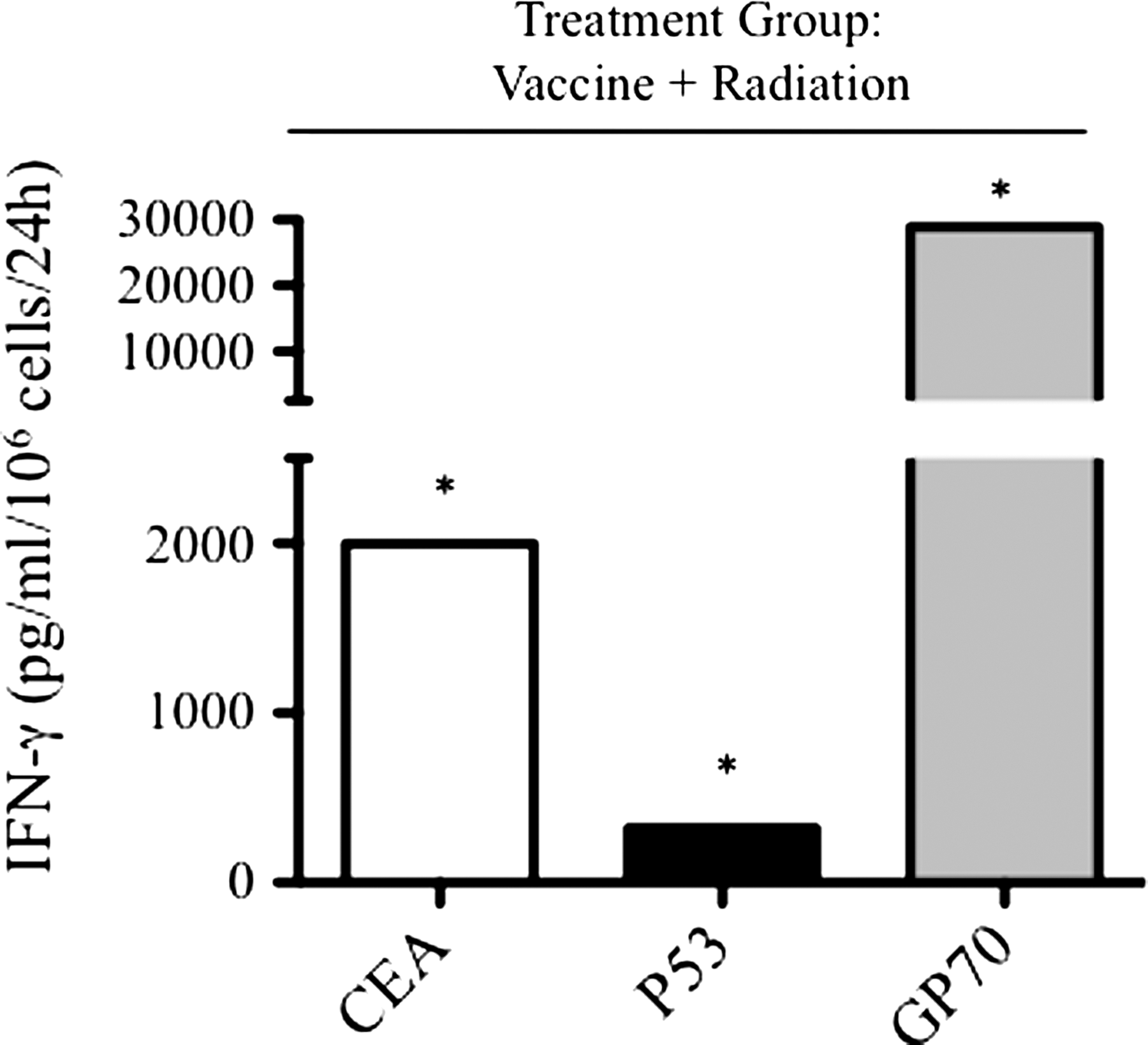
CD8+ T-cell responses to multiple tumor antigens after therapy with vaccine and irradiation. CEA-Tg mice were transplanted with MC38-CEA+ tumor cells. Mice were vaccinated with rV-CEA/TRICOM on day 8 and boosted with rF-CEA/TRICOM on days 15, 22, and 29. Tumors were subjected to radiation (8 Gy) on day 14. Depicted are IFN-γ responses to CD8-restricted peptides specific for CEA (open bars), p53 (black bars), and GP70 (gray bars) from pooled splenic T cells taken from cured mice 6 months after tumor transplant. *: Statistical significance vs age-matched CEA-Tg control mice.
To extend the findings to other potential TAAs, CD8+ T-cell responses specific for MHC class I restricted p53 or GP70 peptides were determined. As before, a significant increase in IFN-γ secreted by CD8+ T cells from mice treated with the combination of vaccine and irradiation was observed in response to p53 or GP70 peptides. Interestingly, in this combination therapy group, the response to GP70 peptide was 15-fold greater than the response to the CEA peptide encoded by the vaccine. T cells from control mice demonstrated no reactivity to the CEA, p53, or GP70 peptides (not shown).
Treatment of a CEA+ tumor with vaccine and radiation mediates abscopal regression of a CEA- tumor at a distal site
The observation that in certain cases radiation treatment of a tumor propagates to tumors outside the irradiated volume is termed “abscopal effect.” 3 It has been suggested that the mechanism for the abscopal effect is in part from an induced systemic immune response. To test this, we designed a tumor model in which mice were transplanted with two tumors in distinct sites. CEA-Tg mice were transplanted with MC38-CEA+ tumors on the right flank and MC38 (CEA-) tumors on the left flank (Fig. 5A). The mice were then divided into those that received (a) no treatment; (b) vaccine alone; (c) irradiation of the MC38-CEA+ tumor alone; or (d) the combination of vaccination followed by radiation of the MC38-CEA+ tumor.
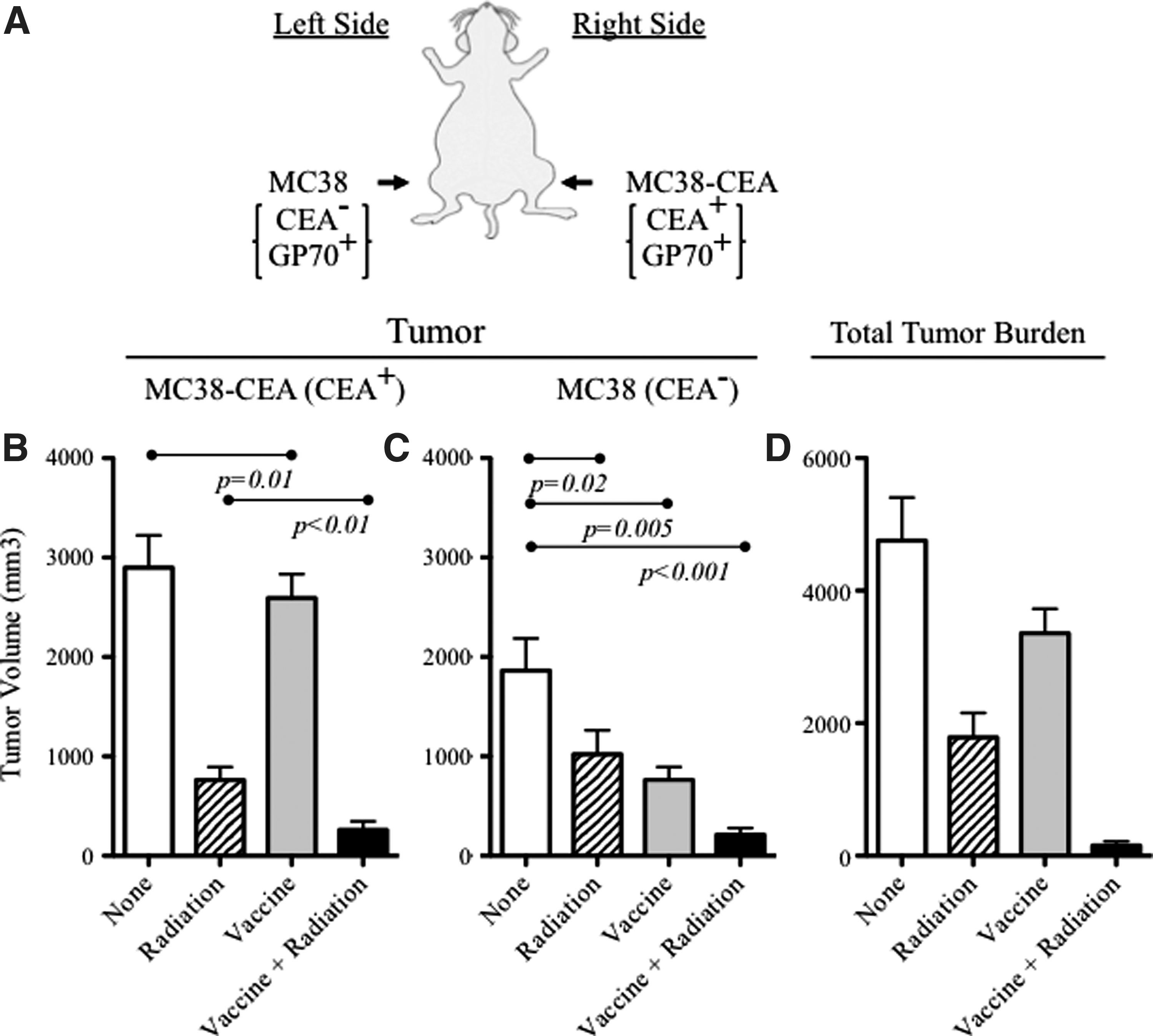
Treatment of a CEA+ tumor with vaccine and irradiation mediates abscopal regression of a CEA- tumor at a distal site. (A) Experiment schema; CEA-Tg mice were transplanted with MC38-CEA+ tumors (subcutaneously right flank) on day 0 and MC38 (CEA-) tumors on the left flank on day 5. Mice were treated with rV-CEA/TRICOM on day 8 and administered 8 Gy external beam radiation to the MC38-CEA+ tumor on day 14. Mice were boosted with rF-CEA/TRICOM on days 15, 22, and 29. All vaccines were given with rF-GM-CSF. Tumor volume on each flank was monitored.
As before, radiation administered at this dose (8 Gy) or vaccine had a modest effect on the treated tumor (Fig. 5B). Therapy of tumors with the combination of the vaccine regimen and irradiation, however, resulted in a significant decrease in tumor growth rate and tumor volume (p=0.001 vs no treatment; p<0.01 vs irradiation alone; p<0.001 vs vaccine alone; Fig. 5B). Strikingly, the combination therapy of vaccine and irradiation of the MC38-CEA+ tumor also mediated significant regression of the MC38 tumor on the opposite side of the mouse (p<0.001 vs no treatment, Fig. 5C). The clinical significance of primary and distal tumor regression was demonstrated by the total tumor burden of these mice (MC38-CEA+ plus MC38; <100 mm3; Fig. 5D).
To extend the observation that combination therapy of vaccine and irradiation initiates a beneficial abscopal effect on nonirradiated tumors, a second mouse model was used. Here mice were transplanted with LL2-CEA+ Lewis lung tumors expressing CEA on the right flank (primary) and at the same time the same tumors (LL2-CEA+) were injected i.v. to form experimental pulmonary metastases. The mice were then divided into those that received (a) no treatment; (b) vaccine alone; (c) radiation therapy of the LL2-CEA+ primary tumor alone; or (d) the combination of vaccination followed by radiation of the LL2-CEA+ primary tumor. Drawing from our observations that GP70 was a potent endogenous tumor antigen (Fig. 4), we used a vaccine encoding this antigen (rV/rF-GP70/TRICOM; Fig. 6A).
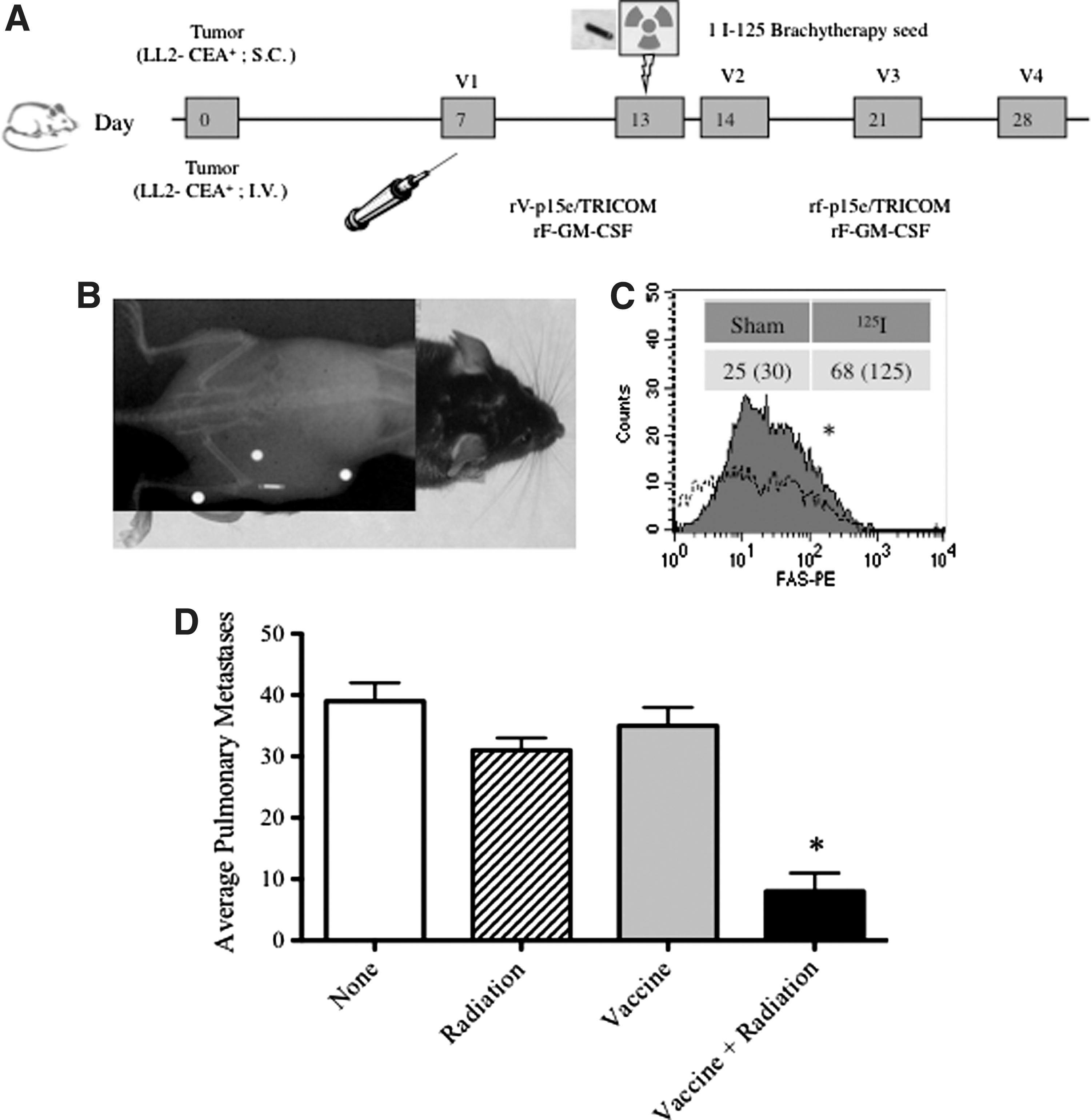
Treatment of a primary tumor with vaccine and irradiation mediates abscopal regression of pulmonary metastases.
Brachytherapy involves implanting a radiation source into the site of a tumor to expose tumor cells to a continuous dose of radiation. To determine if this common form of radiation therapy would mediate similar phenotypic modulation of tumors and subsequent abscopal effects, we used 125I brachytherapy seed implants (Fig. 6B). Similar to external beam radiation, treatment of tumors with 125I brachytherapy resulted in a >two-fold upregulation of Fas (p<0.01 compared with sham treatment, Fig. 6C). Finally, the combination therapy of vaccine and irradiation of the LL2-CEA+ primary tumor also mediated significant regression of the LL2-CEA+ pulmonary metastases (p<0.01 vs all other groups, Fig. 6D).
Discussion
Irradiation is a primary modality in cancer treatment. Irradiation can also reduce tumor growth outside the treatment field, often referred to as the abscopal effect. 23 This effect has been described anecdotally for several types of cancer; however, the mechanisms and therapeutic potential of the abscopal effect have not been fully elucidated. There have been two main hypotheses concerning abscopal tumor regression: (a) Local irradiation induced a release of systemic cytokines that mediate a systemic antitumor effect and/or (b) local irradiation induces systemic tumor specific T-cell responses. There are studies that support each of these nonexclusive hypotheses. In a preclinical model, Camphausen and associates 24 used mice bearing subcutaneous LL2 Lewis lung tumors. These mice were subjected to radiation of the leg (nontumor bearing, five 10 Gy fractions) and subsequently demonstrated significantly slower tumor growth, suggesting that the abscopal effect in this model was not tumor specific. Clinically, Ohba and colleagues 25 showed significant increases in serum TNF-α after local irradiation that was coincident with tumor regression at a distal, nonirradiated site.
These data are in contrast to the tumor models presented here (Figs. 5, 6) in which radiation alone had no discernible effects on distal nonirradiated tumors. These differences might be attributable to the doses of radiation examined. Conversely, in a murine tumor model in which mice were transplanted with CT26 tumors into two anatomic sites (subcutaneous and intrahepatic), treatment of the subcutaneous tumor with fractionated radiation induced antitumor effects on the liver metastases. 26 As the antitumor response of liver tumors was further augmented by the administration of IL-2, this suggested that the mechanism for the abscopal effect was in part from an induced systemic immune response. Our data confirm and extend these implications of immune involvement (Figs. 2, 4, 5, 6).
Furthermore, it has been suggested that irradiation can enhance tumor immunogenicity by promoting dendritic cell cross-priming and the subsequent induction of T-cell responses. Irradiation has been shown to induce inflammatory signals that act as maturation signals for dendritic cells. 27 There, mice with two transplanted mammary carcinoma tumors (67NR) had one tumor treated with external beam radiation (6 Gy). The irradiation impaired the growth of not only the tumor in the radiation field, but also the 67NR tumor outside the treatment area. 27 In that study, Demaria and colleagues 27 also determined that the effect was antigen specific, because in a similar experiment, an unrelated murine B-cell lymphoma (A20) tumor outside the treatment field was not inhibited.
We hypothesized that the abscopal effect encompasses these observations in addition to those described here: Local tumor phenotype modulation and subsequent T-cell killing, cross-presentation of endogenous tumor antigens, and consequent induction of multiple TAA specific CD8+ T-cell populations. Here, we evaluated the potential role of active vaccination in the induction and amplification of irradiation-induced abscopal effects. CEA-Tg mice were vaccinated with a poxvirus-based diversified prime and boost regimen: Recombinant vaccine prime (rV) followed by recombinant fowlpox boosts (rF). These recombinant viruses encoded the TAA gene as well as genes for TRICOM (B7-1, ICAM-1, LFA-3). The advantages of the diversified vaccine prime and boost regimen have been described, 9,28,29 as has the use of T-cell costimulation via inserting multiple costimulatory molecules into these vectors. 7,30,31
It has been reported that irradiation can modulate the phenotype of both murine and human tumor cells, increasing several molecules associated with T-cell recognition and killing. 32 –34 Here, we observed upregulation of the death receptor Fas after radiation exposure in two different tumor cell types (Figs. 1, 6). Interestingly, the relative levels of Fas upregulation were similar from either 8 Gy external beam (Fig. 1) or a 125I brachytherapy seed implant (∼29 Gy total delivered dose; Fig. 6). Fas upregulation has been previously determined in a mouse model to be responsible for increased sensitivity to T-cell killing after irradiation. 32
In many human tumor cells, Fas is defective in the signaling cascade required for cell death. These cells, however, upregulate other molecules involved in T-cell recognition, including MHC-I, ICAM-1, and tumor antigens. 33 Garnett and associates 33 demonstrated that irradiation was able to alter the cell-surface expression of a variety of immunomodulatory molecules such as Fas, ICAM-1, MHC-I, and TAAs such as CEA and mucin-1 (MUC-1). Multiple human carcinoma cell lines (n=17) were examined for responses to nonlethal doses of radiation (10 or 20 Gy) and found that at least one of the above-named surface molecules increased in 21 of 23 (91%) cell lines studied. Furthermore, all irradiated cell lines demonstrated significantly enhanced killing compared with nonirradiated cell lines, suggesting that nonlethal doses of radiation render human tumor cells more amenable to immune recognition and attack.
We sought to induce CEA-specific T cells via active specific immunotherapy; priming CEA-Tg mice with rV-CEA/TRICOM and boosting with rF-CEA/TRICOM, and combining this modality with external-beam radiation (8 Gy). This dose and schedule of radiation was determined to be noncurative; the MC38-CEA+ tumors exhibited a marginal, but insignificant reduction in growth, compared with nonirradiated tumors (Fig. 2C). The vaccine regimen alone did not affect the growth rate of the transplanted tumor. The combination of the vaccine regimen with irradiation in the 8-day model resulted in a significant reduction of tumor volume. In addition, 40% of the mice in this treatment group had a completely eradicated tumor mass (Fig. 2E).
These data are similar to the data previously described for the combination of low-dose external beam radiation therapy and active therapeutic vaccination for the treatment of subcutaneous tumors in a mouse model. 32 MC38-CEA+ tumor destruction in response to the combination of the vaccine regimen with irradiation was shown to be associated with active vessel remodeling and massive infiltration of CD3+ T cells, which was confirmed by immunohistochemistry (Fig. 3). It has been demonstrated that irradiation alone can upregulate the vascular endothelial marker CD31 35,36 and induce a remodeling of the tumor vasculature in the microenvironment. 37 The data shown here (Fig. 2) are consistent with those reported by Quarmby and colleagues, 36 and Ganss and colleagues, 38 in which whole-body lethal irradiation resulted in upregulation of CD31 and remodeling of the tumor vasculature, permitting T-cell access and subsequent tumor therapy.
Antigens from peripheral tumor cells can enter the MHC class I pathway for presentation by host APCs to CD8+ T cells via a process commonly described as “cross-presentation.” 39 It has been suggested that therapy protocols using damage-inducing agents could prime antitumor immune responses. Antigen cascade had been previously described as an immunologic response demonstrating epitopes distinct from and non–cross-reactive with the inducing epitope. Other similar observations have been termed epitope spreading, determinant spreading, cryptic determinants, or antigen spreading. Antigen cascade could be a possible mechanism for the regression of antigen variant tumors, tumors at distal sites, and micrometastatic deposits. MC38-CEA+ tumor cells, in addition to expressing CEA, have been noted to overexpress other TAAs such as wild-type p53 11 and endogenous germline retroviral encoded proteins, including GP70. 13,22
Here, it was shown that mice cured of MC38-CEA+ tumors by the combination of the vaccine regimen and irradiation generated CD8+ T-cell responses not only against CEA, but also against p53 and GP70 (Fig. 4). Because p53 and GP70 were not encoded by the vaccine regimen, it can be inferred that these antigens were donated from the tumor itself. Interestingly, the IFN-γ response against GP70 was 15-fold greater than that against the CEA peptide, and it can be postulated that the majority of the antitumor effect was mediated by GP70 responses.
Similar to the observed cascade response to endogenous tumor antigens (Fig. 4), antigen cascade has been observed in PBMC samples taken from patients receiving vaccine and radiation therapy. 40 A recent clinical trial assessed the use of a recombinant poxviral-based vaccine expressing prostate-specific antigen (PSA) combined with standard definitive radiotherapy in patients with localized prostate cancer. 40 There, T-cell responses specific for at least one additional endogenous TAA after therapy developed in six of eight patients. These included the generation of T cells against PSMA, PAP, PSCA, and/or MUC-1.
Also similar to our data (Fig. 4), in some cases the immune response to a cascade antigen was more prominent than the immune response directed against the antigen encoded by the vaccine. Further supporting these observations, Nesslinger and associates 41 reported that patients subjected to external beam radiation or brachytherapy for prostate cancer developed de novo antibody responses to additional prostate antigens after exposure, while patients receiving radical prostatectomy did not.
Our hypothesis was that the antigenic cascade was in part responsible for the abscopal effect. To test this, an experimental model was developed where mice were transplanted with an antigen defined primary tumor, MC38-CEA+, which endogenously expresses CEA, p53, and GP70; and a distal tumor, MC38, which expresses only p53 and GP70. In this model, mice were vaccinated with rV/rF-CEA-TRICOM, and the primary tumor was treated with irradiation (Fig. 5). Mice demonstrated significant tumor regression of both the irradiated and nonirradiated tumors, presumably via T-cell responses to p53 and GP70 (Fig. 4). The antigen cascade mediated abscopal effect was further confirmed in a different animal model bearing both a subcutaneous tumor and pulmonary metastases. There, systemic vaccination and treatment of the primary tumor via 125I-brachytherapy seed implant mediated regression of the distant pulmonary metastases (Fig. 6).
Results from these and other preclinical studies provided the rationale for additional clinical evaluation of the combination of radiation therapy and therapeutic cancer vaccines. One clinical trial assessed the use of a recombinant poxviral-based vaccine expressing PSA combined with standard definitive radiotherapy. 40 Results from this clinical trial indicated that the combination was safe, well tolerated, and, more importantly, effective at generating PSA-specific immune responses. Approximately 76.5% of patients in the combination therapy arm showed a ≥three-fold increase in PSA-specific T cells vs 0% in the radiation-alone arm (P<0.0005). 40 Another ongoing clinical trial is combining the rV/rF-CEA/TRICOM vaccine with low-dose external beam radiation delivered directly to liver metastases in patients with CEA+ solid tumors. 42 An additional radiation modality, Samarium-153 (153Sm-EDTMP; Quadramet®, Cytogen, Princeton, NJ), is currently being studied in a randomized phase II trial to determine if 153Sm-EDTMP combined with an rV/rF-PSA/TRICOM vaccine (PROSTVAC®, Bavarian Nordic (BN) Immunotherapeutics, Mountain View, CA) can improve time to progression over 153Sm- EDTMP alone in patients with castrate-resistant prostate cancer (CRPC) metastatic to bone. 43
Other emerging immunotherapies such as the vaccine Sipuleucel-T (Provenge®, Dendreon Corp, Seattle, WA) or anti-CTLA-4 (Ipilimumab (Yervoy™), Bristol-Myers Squibb, Princeton, NJ) might also be able to be added to radiation therapy to exploit the abscopal effect. Indeed, the combination of anti-CTLA-4 and fractionated radiation therapy was observed to induce an abscopal effect in a murine model. 44
Many foundational biologic events have been hypothesized for the abscopal effect mechanism of action, including (a) the possible role of the immune system, 2,23 (b) inflammatory mediators and dendritic cell activation, 23,24,27 and now (c) induction of antigenic cascade (Fig. 7). Exposure of cells to radiation is an immunologically active process wherein damaged tumor cells release TAAs that can potentially be exploited to stimulate robust tumor-specific immune responses (Figs. 2, 4, 5, 6). Cells undergoing irradiation treatment develop distinctive changes on their plasma membranes, which act as danger signals such as Fas to promote T-cell recognition and killing (Figs. 1, 6).
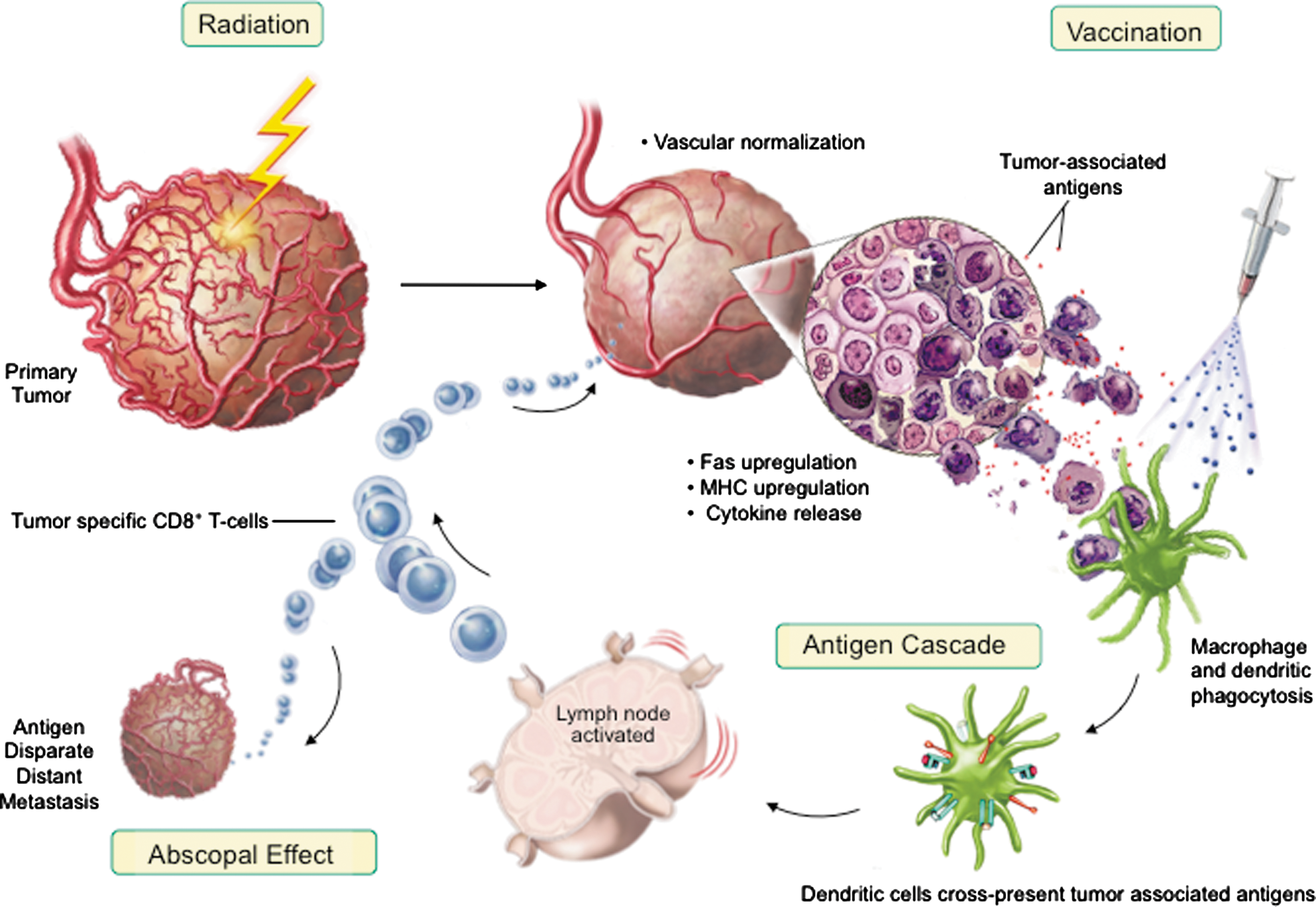
Model of antigen cascade mediated abscopal effect.
Friedman 45 has previously described a “danger model” of immunity wherein ionizing radiation generates an inflammatory microenvironment filled with apoptotic and necrotic cells, cytokines, chemokines, inflammatory mediators, and acute-phase reactant proteins. This milieu of immune modulators can activate APCs and support their processing of newly exposed TAAs. Activated APCs then migrate to the location of radiation-induced cell death, undergo maturation, and present postradiation cellular debris and antigens to T cells. These additional tumor antigens are now able to participate in the immune reaction (Fig. 4), cycling back to both the irradiated tumor and other tumors outside the radiation field (Figs. 5, 6). This antigenic cascade ultimately can mediate abscopal regression of antigen disparate distal tumors (Fig. 7).
It has been suggested that certain chemotherapeutic regimens trigger cancer cell death while stimulating immune responses against the tumor. This immunogenic cell death and subsequent immune responses have been shown to be critical for the clearing of tumor cells that survive therapy. 46,47 Here, we suggest that irradiation may play a similar role. Although local control of the primary tumor is necessary and can usually prevent metastasis, irradiation generally fails to control preexisting systemic disease, which may be present as undetectable micrometastases. By coupling tumor irradiation with immunotherapy induced polyclonal T-cell responses (antigen cascade), however, the abscopal effect can transcend from anecdotal observation to a defined mechanism that can be exploited for the treatment of systemic disease.
Footnotes
Acknowledgements
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health (NIH).
The authors thank Marion Taylor for excellent technical assistance, and Debra Weingarten for her editorial assistance in the preparation of this manuscript. We thank Dr. Kevin Camphausen, NCI, NIH, for assistance with radiation administration, and Dr. Alfredo Molinolo, NIDCR, NIH, for digital photomicroscopy. We thank Dr. Jeffrey Schlom, NCI, NIH, for his helpful suggestions in the review of this manuscript.
Disclosure Statement
No competing financial interests exist.