
Abstract
miR-34a was identified as one of the downregulated microRNAs (miRNAs) in human lung cancer. However, the precise biological role of miR-34a in p53 deficient lung cancer cell lines remains largely elusive. In the present study, we aimed to identify the role of miR-34a in the regulation of lung cancer cell proliferation. Using quantitative RT-PCR analysis, we found that miR-34a was highly upregulated in the p53 wild-type A549 human lung cancer cell line when treated with the DNA damaging agent adriamycin (ADR), but not in the SBC-5 cells harboring mutated p53. Transient introduction of miR-34a into A549 and SBC-5 cell lines caused complete suppression of cell proliferation and induced the cell cycle arrested at the G1 phase. When we knockdown the miR-34a downstream target—Sitr1— using the small-interfering RNA, there was also a cell growth inhibition in both cell lines though not as much as miR-34a did. Moreover, we demonstrated that pretransfection of miR-34a could increase the sensitivity of both lung cancer cell lines to cisplatin (DDP), and this could be reverted by the miR-34a inhibitor. Moreover, when cells pretreated with siR-Sirt1, they are more sensitive to DDP than the control pretreated cells as well. We thus hypothesize the miR-34a/Sirt1 cascade involved with p53-independent functions. Overall, in this study, we found the proliferation inhibition function of miR-34a in vitro in lung cancer cell lines is p53 independent, and also demonstrated the combination therapeutic potential of miR-34a and DDP in lung cancer cell lines.
Introduction
MicroRNAs (miRNA) are a class of small (18–25 nucleotides) noncoding RNA molecules that modulate gene expression post-transcriptionally. 1 –3 Bioinformatics estimates suggest that more than 60% of the human genome may be regulated by miRNAs. 4 miRNAs can affect various signaling pathways 5 and cellular events, such as cell cycle distribution, differentiation, and apoptosis. 6 –8 No wonder, dysregulation of this small molecule can contribute to the development of human diseases, including cancer, 8 and miRNAs, therefore have the potential to be the cancer therapeutic target. 9,10 miRNAs can act as both oncogenes and tumor suppressor genes. Among the miRNAs that play the role of tumor suppressor in various human cancers, miR-34a is one of them. 11 –19 miR-34a is located on chromosome 1p36.22, and was first found as a potential tumor suppressor in neuroblastoma, which is one of the most common cancers in children. The reduced expression level of miR-34a is also linked to many other human cancers, including prostate, pancreatic cancer cells, colon cancer specimens, and lung. Researches demonstrated that miR-34a expression can be induced by the tumor suppressor p53, 11 once p53 is activated, miR-34a production is increased. Furthermore, miR-34a can also form a positive feedback loop with p53 expression through downregulation of its targeted gene, Sirt1. 13,20 The miR-34a-induced decrease of Sirt1 expression permits an upregulation of acetylated p53 and an increase of p53 activity. Increased p53 activity on the other hand can enhance miR-34a production, thus completing the positive feedback loop. Exotic expression of miR-34a inhibits the growth of cultured cancer cells, induces a senescence-like phenotype, and could also lead to apoptosis and inhibition of migration. 17 Studies also showed that hypermethylation of the miR-34a promoter can also lead to reduced miR-34a expression in lung cancer as well as other cancer types. 21 In a word, miR-34a displays a tumor suppressor function in numerous cancer cells. However, the in vitro study of antioncogenic activity of miR-34a in lung cancer are all performed in p53 wild-type cell lines, and its potential as a therapeutic target in p53 deficient cell lines remains unknown. Here in the present study, we found that the proliferation inhibition function of miR-34a in vitro in lung cancer cell lines is p53 independent, and then demonstrated the combination therapeutic potential of miR-34a and DDP in lung cancer cell lines.
Materials and Methods
Cell lines and adriamycin treatment
Human lung adenocarcinoma cell line, A549 and SBC5 cells were purchased from American Type Culture Collection and cultured in the Dulbecco's modified Eagle's medium with fetal bovine serum (FBS; Gibco BRL). Media were supplemented with 10% FBS, 50 units/mL of penicillin, and 50 μg/mL of streptomycin. The cells were routinely incubated at 37°C in a humidified atmosphere with 5% CO2. Adriamycin (ADR) was purchased from Sigma-Aldrich. For the time course experiment of ADR treatment, A549 cells were seeded at 6×105 cells per well in six-well plates 1 day before treatment, and then incubated with or without ADR in the culture medium. Cells were collected and subjected to extraction of total RNA and protein at 0, 2, 4, 8, 12, 24, and 48 hours. For isolation of total RNA and protein, cells were seeded into six-well plates, and then treated with ADR at a concentration of 100 ng/mL for 24 hours.
Transfection of cells with miRNA and inhibitors
Cells were seeded into six-well plates at a density of 5×105 cells per well 24 hours before transfection. A549 and SBC5 cells were transfected by using the lipofectamine2000 transfection reagent (invitrogen) following the manufacturer's instructions. For each transfection, 10 pmol of precursor miRNA oligonucleotides or inhibitors (Genepharm) in 250 μL of Opti-MEM (Invitrogen) was mixed with 250 μL of Opti-MEM that contained 5 μL of lipofectamine2000 reagent, which had been preincubated for 5 minutes at room temperature. The mixture was incubated for 20 minutes at room temperature, and then added to the cells. The cells were harvested 24 hours after transfection for flow cytometry and Western blot analysis.
Cell cycle analysis
The effect of miR-34a on cell cycle distribution was determined by flow cytometry using the cell cycle detection kit (Keygen). Briefly, 24 hours post-transfection, adherent cells were collected, washed twice with phosphate-buffered saline (PBS), then resuspended in 0.2 mL PBS, then added the solutions according to the manufacturer's protocol. Before analysis, cells were resuspended, and then analyzed by flow cytometry. The relative proportions of cells in the G1, S, and G2/M phases of the cell cycle were determined by the flow cytometry data.
Western blotting
Cells were harvested and suspended in the lysis buffer (1 mM dithiothreitol, 0.125 mM EDTA, 5% glycerol, 1 mM phenylmethylsulfonylfluoride, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 1 μg/mL aprotinin, 1% Triton X-100 in 12.5 mM Tris-HCl buffer, pH 7.0) on ice. The cell extracts were clarified by centrifugation and the protein concentrations were determined by using the Bio-Rad protein assay kit. Each protein extract (15 μg) was electrophoresed on a 10% sodium dodecyl sulfate–polyacrylamide gel, transferred to the PDVF membrane in a buffer containing 25 mM Tris-HCl (pH 8.3), 192 mM glycine, and 20% (v/v) methanol, and blocked in 5% (w/v) nonfat milk in Tris-buffered saline-Tween 20 (0.1% by volume, TBST) for 2 hours at room temperature. The membrane was incubated overnight at 4°C with primary antibodies for p53 and β-actin (Santa Cruz Biotechnology). Primary antibodies were removed and the blots were extensively washed with TBST for three times. Blots were then incubated for 1 hour at room temperature with the secondary antibody (goat anti-mouse antibody coupled to horseradish peroxidase, 1:5000 dilution). Following removal of the secondary antibody, blots were extensively washed as above for 1 hour and visualized by using the chemiluminescence-enhanced chemiluminescence kit (Santa Cruz), and the specific bands were recorded on X-ray film.
3-(4, 5-Dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay
Cell proliferation was determined by the 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma) assay as described elsewhere. 22 Briefly, the cells were plated in 96-well tissue culture plates at a density of 1×104 cells per well and allowed to attach overnight; cells were transfected with miR-34a, si-Sirt1, or an inhibitor for 24 hours, and then treated with different concentrations of DDP for 48 hours, then incubated with MTT (20 μL of 5 mg/mL) for 4 hours. The formozan precipitate was dissolved in 150 μL dimethylsulfoxide and the absorbance at 570 nm was measured by a benchmark microplate reader (Bio-Rad).
Results
Induction of the miR-34a expression in response to ADR in human lung cancer A549 cells
A549 lung cancer cells were treated with 5 μg/mL of ADR for different time points, the expression of miR-34a and p53 were detected. Figure 1 shows that the expression of both, the p53 and miR-34a, was increased in a time-dependent manner after ADR treatment. The expression of miR-34a rose 2.5-fold at 8 hours and 7-fold at 48 hours (Fig. 1A). We also observed that p53 started to accumulate at 2 hours, and the accumulation continued until 48 hours after treatment (Fig. 1A). SBC-5 cells showed no change in expression of miR-34a after ADR treatment (Fig. 1B). These results indicate that miR-34a is induced in a p53-dependent manner after ADR treatment.

Induction of miR-34a and P53 expression post-treatment with ADR in lung cancer cell lines harboring wild-type p53.
MiR-34a contributes to the cell growth inhibition and cell cycle arrest of lung cancer cell line
We then examined the effect of miR-34a on proliferation of A549 and SBC5 cell line. A549 and SBC5 lung cancer cells were transfected with 10 pmol of miR-34a for 48 hours and cell viability was then analyzed by MTT assay. The introduction of miR-34a caused a remarkable inhibition of cell proliferation in both A549 and SBC5 cells compared with that of control miRNA. Especially in SBC-5 cells, the proliferation rate was reduced nearly 60% compared with the control miRNA transfected group (Fig. 2A).
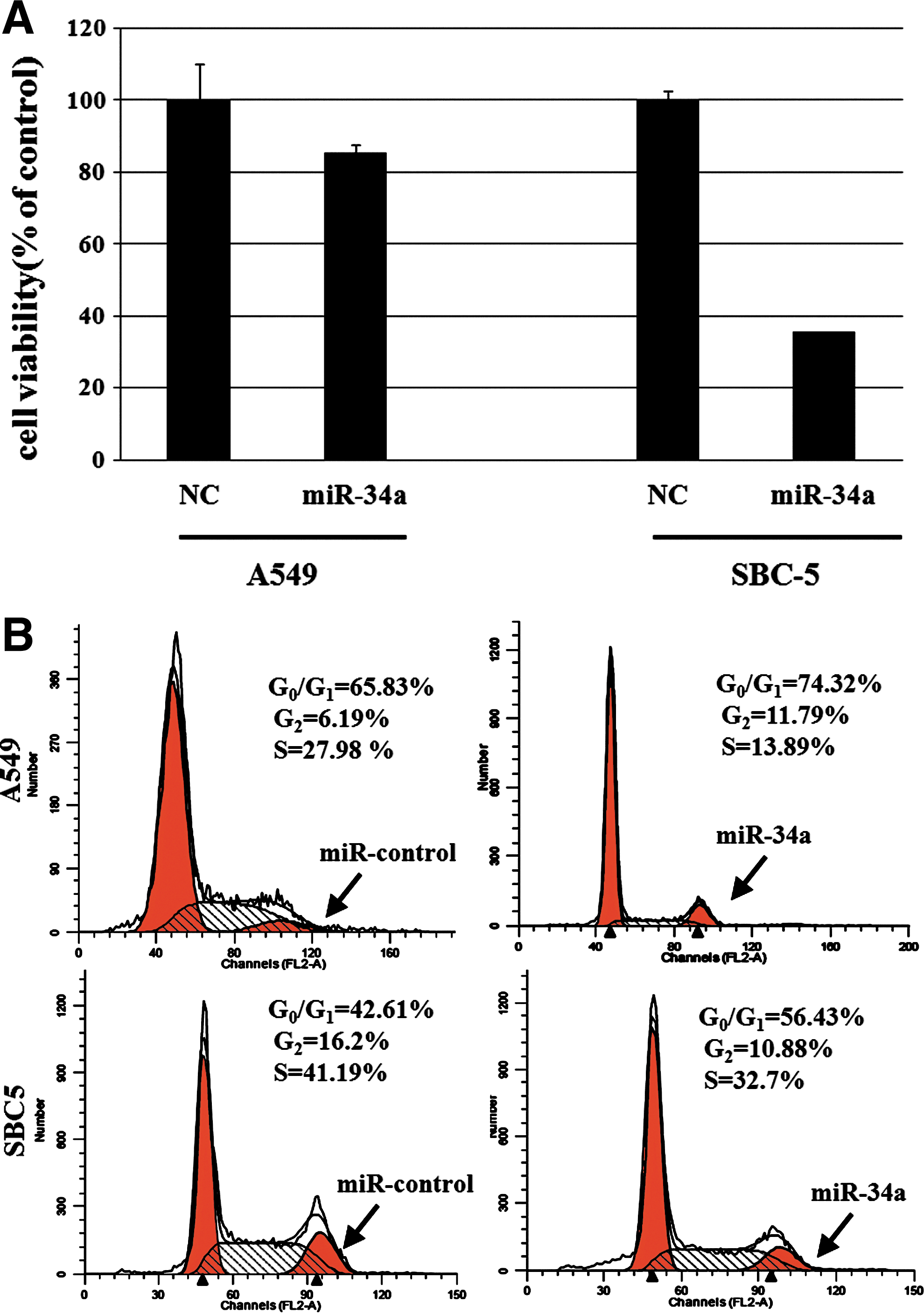
Effects of miR34a on cell growth and cell cycle arrest in lung cancer cell lines.
The effects of miR-34a on cell cycle progression were investigated using flow cytometry. A549 and SBC5 lung cancer cells were transfected with 10 pmol of miR34a for 24 hours, and then analyzed for cell cycle distribution by means of flow cytometry. Overexpression of miR34a increased the population of cells in the G0-G1 phase with a reduction of cells in the S phase. As shown in Figure 2B, in A549 cells, the G1 phase population was increased from 65.8% to 74.3%, and the result also showed that in the p53-mutated SBC5 cell line, the G1 phase population was increased from 42.6% to 56.4%. The data are represented as mean±standard deviation from at least three independent experiments, and indicated that miR34a led to cell cycle arrest at the G1 phase in both cell lines.
Small-interfering RNA targeted Sirt1 inhibited cell growth in lung cancer cell lines
As Sirt1 was the downstream target of miR-34a, we also observed the reduced expression of Sirt1 post-miR-34a transfection in both cell lines (Fig. 3A). We then designed an experiment to evaluate the effect of Sirt1 knockdown on the proliferation of lung cancer cells. A549 and SBC5 cells were seeded in 6-well plates at a concentration of 2×105/well 1 day before transfection. The small-interfering RNA (siRNA) for Sirt1 was used for transfection using lipofectamine2000, according to the manufacturer's protocol. The sequence of siRNA for Sirt1 was ACAAUGAGGAGGUCAACUUC. Stable negative control siRNA (GenePharma) was used as a control. The effects manifested by the introduction of siRNA into the cells were assayed at 48 hours post-transfection. Figure 3B showed that the expression level of Sirt1 gene in lung cancer cells was significantly reduced post-siRNA transfection. We then examined the effect of Sirt1 inhibition on the proliferation of A549 and SBC5 cell lines. A549 and SBC5 lung cancer cells were transfected with 10 pmol of siRNA for 48 hours and cell viability was then analyzed by MTT assay. The inhibition of Sirt1 caused reduced cell proliferation in both A549 and SBC5 cells compared with the control miRNA transfected group, though not as much as the miR-34a transfected groups compared to Figure 2B.

Effects of Sirt1 knockdown on A549 and SBC-5 cells.
miR-34a sensitizes lung cancer cell lines to DDP independent of p53 status
As DDP was the standard chemotherapy reagent in lung cancer treatment, we hypothesize if the miR-34a expression has any effect on DDP sensitivity in A549 and SBC5 cells. We transfected A549 and SBC5 cells with miR-34a for 24 hours at 10 pmol, which concentration was determined from the previous data, and then treated the cells with different concentrations of DDP for another 48 hours, and cell viability was then analyzed by MTT assay. In this experiment, the pretreatment with miR-34a before DDP exposure increased the sensitivity to DDP in lung cancer cells at all concentrations of DDP examined compared with the NC pretreatment group (Fig. 4).
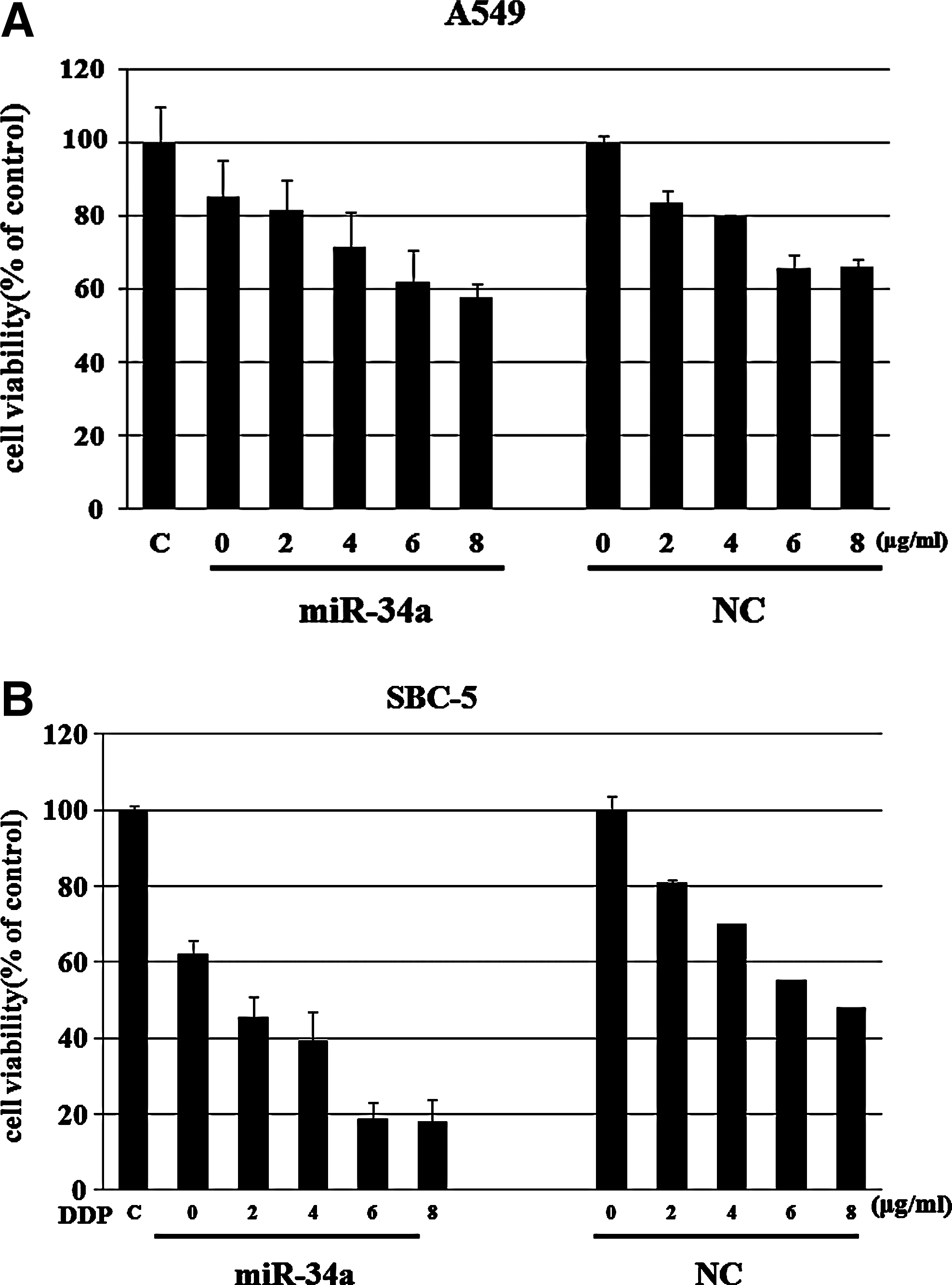
Effects of ectopic expression of miR-34a on the sensitivity of DDP in lung cancer cells.
To investigate the relationship between the upregulation of miR-34a and sensitivity to DDP, we pretransfected the A549 and SBC5 cells with miR-34a inhibitor before DDP treatment. Interestingly, pretreatment with the inhibitor efficiently prevented the growth inhibition by DDP (Fig. 5), in both cell lines. As this increase in miR-34a expression after DDP treatment of the parental cells was significantly suppressed by the miR-34a inhibitor, these results clearly demonstrated that the increase of the miR-34a level determined the sensitivity to DDP in the parental cells.

Effects of miR-34a inhibitor efficiently prevents the growth inhibition induced by DDP in A549 and SBC-5 lung cancer cells.
Discussion
Recent data showed that human carcinogenesis could be influenced by the altered expression of miRNA. 23 miR-34a has been reported to be downregulated in a number of cancers, such as pancreatic, 24 colorectal, 25 breast, 26 lung, 27,28 prostate, 29,30 bladder, 31 and leukemia. 32 miR-34a expression was transcript regulated by the tumor suppressor p53, which is also discovered in our study.
In the present study, miR-34a was identified as a DNA damage-responsive gene in the A549 lung cancer cell line after treatment with the DNA damage drug ADR. In contrast, no obvious change of the expression level of miR-34a was observed in the SBC-5 cells harboring mutant p53. Ectopic expression of miR-34a by transfection with mature miR-34a inhibited cell growth not only in the A549 cells, but also inhibited the growth of SBC-5 cells significantly. Seventy-two (72) hours post-transfection, the proliferation rate reduced 20% in A549 cells; interestingly, the proliferation of SBC-5 cells was inhibited to 62%. At the mean time, the expression of miR-34a downstream target Sirt1 was also reduced in both cell lines. When we knockdown Sirt1 expression by RNA interference, the proliferation rates of both cell lines was also reduced, though not as much as the miR-34a did. This led us to hypothesize that the miR-34a can affect lung cancer cell proliferation independent of P53 status. We then designed an experiment to detect the influence of miR-34a on the sensitivity of lung cancer cells to DDP, the standard chemotherapy reagent of lung cancer. Compared with NC pretreated cells, the pretreatment with miR-34a before DDP stimulation increased the sensitivity to DDP in both A549 and SBC-5 lung cancer cells at all of the concentrations of DDP we have examined.
Yukihiro Akao et al. 33 demonstrated a significant contribution of the miR-34a/Sirt1 cascade to chemoresistance in human prostate cancer PC3 cells and colorectal cancer DLD/5-FU cells. In the present study, we found that when pretreated, the A549 and SBC-5 lung cancer cells with miR-34a, the sensitivity of both cell lines to DDP was increased, and this could be reverted by the miR-34a inhibitor. To evaluate the contribution of miR-34a/Sirt1 cascade to the sensitivity of DDP, we then designed the siR-Sirt1 to inhibit the expression of Sirt1. The cell growth was also inhibited through knockdown of Sirt1 by siR-Sirt though not as much as the miR-34a did; moreover, cells pretreated with siR-Sirt1 are more sensitive to DDP than the control pretreated cells as well. We thus hypothesize the miR-34a/Sirt1 cascade or other miR-34a-associated cascades involved with p53-independent functions; further studies to clarify our hypothesis and the detailed molecular mechanism are needed. In summary, our present data provide strong evidence for the therapeutic potential of miR-34a in the treatment of lung cancer.
Footnotes
Disclosure Statement
The authors declare that no conflict of interest exists in the submission of this manuscript.