
Abstract
Reprogramming of cancer cells into induced pluripotent stem cells (iPSCs) opens up the possibility of converting malignant cells into any cell type, including those best suited to be developed as cancer vaccines. Mouse models are needed to evaluate and optimize the therapeutic efficacy of such novel cancer vaccines. However, only human cancer cell lines have been reported as being reprogrammed into iPSCs. Here, we report a proof-of-principle study which shows that mouse cancer cells can be reprogrammed into iPSCs that are capable of subsequent differentiation. Four canonical reprogramming transcription factors, Oct3/4, Sox2, Klf4, and c-Myc, were introduced by plasmid transfection into mouse Lewis lung carcinoma D122 harboring Nanog-GFP reporter. Green fluorescent cells were found clustered into embryonic stem cell (ESC)-like colonies expressing ESC markers, Oct4 and SSEA-1. Bisulfite genomic sequencing analyses of these cells revealed hypomethylation of the Nanog promoter. The expression of a host of pluripotency genes by these reprogrammed cells at levels similar to those of ESCs was confirmed by quantitative real-time PCR. Functional pluripotency of the reprogrammed cells was demonstrated by their ability to form embryoid bodies and differentiate into neuronal progenitors on retinoic acid treatment. This study indicates the feasibility of developing iPSC-based experimental cancer vaccines for immunotherapy in mouse models.
Introduction
Induction of pluripotent stem cells from mouse embryonic and adult fibroblasts by introduction of four transcription factors, Oct3/4, Sox2, Klf4, and c-Myc, also known as Yamanaka factors, was first reported in 2006. These induced pluripotent stem cells (iPSCs) were able to form cell types of all three embryonic germ layers in embryoid bodies (EBs) and teratomas, as well as to contribute to early mouse embryonic development when injected into blastocysts. 1 Since then, a wide variety of mouse and adult human somatic cell types, including primary cells and nontransformed cell lines, have been found amenable to reprogramming into iPSCs using Yamanaka factors or similar combinations introduced by viral or nonviral vectors, as synthetic RNAs, or recombinant proteins. 2,3 The advent of iPSC technology has opened up the possibility of generating patient-specific stem cell lines, which can then be directed to differentiate into specific cell or tissue types for pathological investigations and therapeutic applications. 4 It has been shown, for example, that iPSCs obtained by the reprogramming of normal human and mouse fibroblasts can be directed to differentiate into dendritic cells, a potent antigen-presenting cell type that is commonly employed in generating therapeutic cancer vaccines. 5 –8 Recently, the reprogramming of human cancer cell lines has been explored as an approach for studying the interaction of cancer-related genes with cell environment and differentiation, as well as for discovering novel cancer treatments. 9 –11 These studies demonstrated that reprogrammed human cancer cells also possess the pluripotency which is capable of differentiation into multiple cell lineages of all three germ layers. Thus, converting cancer cells into highly immunogenic tumor antigen-presenting dendritic cells for cancer immunotherapy has become a distinct possibility. Similar to any novel therapeutic treatments under development, this promising possibility should first be tested in mouse models. Since only human cancer cell lines have hitherto been shown to be reprogrammable, 9 –11 we sought to investigate whether mouse cancer cells can also be reprogrammed into iPSCs that are capable of directed differentiation into a specific cell type. Our study showed that mouse iPS-like cells can indeed be generated by the reprogramming of mouse Lewis lung carcinoma D122, which, in turn, can be further be directed to differentiate into another cell type, neuronal precursors, indicating that it may be possible to generate mouse cancer vaccine models based on iPSC technology.
Materials and Methods
Plasmids and cell lines
A reprogramming plasmid pCAG2LMKOSimO 12 encoding all the four Yamanaka factors, Oct3/4, Sox2, Klf4, and c-Myc (OSKM), in a single transcript was a gift of Dr. Keisuke Kaji (Addgene plasmids #20866). D12213 and mouse ES cells CCB 14 are generous gifts from Dr. Lea Eisenbach at the Department of Immunology of the Weizmann Institute of Science, Rehovot, Israel, and Dr. Michael Neuberger of MRC Laboratory of Molecular Biology, Cambridge, UK, respectively. The G418-resistant and LIF-secreting immortalized mouse fibroblasts, SNL Feeder Cells, were purchased from Cell Biolabs. D122 and SNL were maintained in DMEM supplemented with 10% FBS. The CCB cell line was maintained in ES medium (ESM) comprising Knockout™ DMEM (Invitrogen) supplemented with 15% FBS, 1 mM MEM nonessential amino acids solution (Invitrogen),1 mM L-glutamine (Invitrogen), 0.1 mM 2-mercaptoethanol (EmbryoMax ES Cell Qualified; Millipore), and LIF (1000 U/mL; Millipore).
Generation of D122 containing mouse Nanog-GFP promoter reporter
Mouse Nanog Stem Cell Differentiation Reporter lentiviral particles (System Biosciences) were used to transduce D122 according to the manufacturer's protocol. Briefly, on day 0, D122 cells at 1×105 were transduced with the lentiviral particles at MOI of 5 in DMEM medium supplemented with 8 μg/mL polybrene for 3 days. The cells were then cloned and used for reprogramming.
Induction of iPSCs from D122
D122 cells containing the Nanog-GFP promoter reporter were transfected at 5×105 per well with 3 μg of PvuI- linearized pCAG2LMKOSimO on day 0 by nucleofection using nucleofector solution V and program T-30 of nucleofector® II Device (Lonza). The transfected cells were cultured in DMEM supplemented with 10% FBS (ES-qualified; Invitrogen) (DM) for the first 2 days. For the next 7 days, the culture medium was changed to ESM with the addition of 2 mg/mL G418 (Invitrogen). On day 9, the cells were trypsinized and plated on mitomycin-treated SNL feeder cells (50,000 cells/cm2) (Cell BioLabs) in LIF-supplemented ESGRO complete medium (Millipore) containing 2 mg/mL G418. On day 20, colonies with green fluorescence were picked manually into 1% gelatin-coated plate containing LIF-supplemented ESGRO medium for expansion. Cells were passaged 1:10 every 3 days using accutase (Millipore) in place of trypsin.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for 15 minutes at 25°C, washed thrice with phosphate-buffered saline (PBS), and blocked in PBS containing 3% FBS for 20 minutes and 0.1% Triton X-100 (Sigma-Aldrich). Primary antibodies against Oct4 (1:100; Stemgent), Nanog (1:100; Stemgent), PAX6 (supernatant, 1:100; Developmental Studies Hybridoma Bank, University of Iowa), or Nestin (supernatant, 1:100; Developmental Studies Hybridoma Bank) in PBS containing 3% FBS and 0.1% Triton X-100 were applied to the cells for 1 hour at 25°C. After three washes in PBS, appropriate secondary antibody, Alexa Fluor 568 rabbit antimouse IgG (1:100; Invitrogen) or Alexa Fluor 568 donkey antirabbit IgG (1:100, Invitrogen), was applied to the cells for 1 hour at 25°C, and visualized using an epifluorescence microscope (Lecia DM IRB).
Quantitative real-time PCR
Total RNA was prepared using an RNeasy kit (Qiagen), and reverse transcribed using RNA superscript III reverse transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed using the Applied Biosystem 7900HT System with SYBR Green-based PCR Master Mix (Roche). The PCR primer sequences for the pluripotency genes were according to Kaji et al. 12 Mouse TBP gene expression was used for normalization, and the ΔΔCt method was employed to calculate the gene expression levels relative to those in CCB mouse ES cells.
Bisulfite genomic sequencing
Bisulfite treatment was performed using the EpiTect Bisulfite Kit (Qiagen) according to the manufacturer's instructions. The Nanog promoter region of the bisulfite-converted DNA was amplified by PCR and cloned into pCR™4-TOPO® TA vector (Invitrogen) for DNA sequencing by BGI-Hongkong Co., Limited using M13 forward and reverse primers. The PCR primers for mouse Nanog promoter were: Forward 5′-gattttgtaggtgggattaattgtgaattt, Reverse 5′-accaaaaaaacccacactcatatcaatata. 1
EB formation and neural cell differentiation
For EB formation, 500 D122-iPS-1C cells in 20 μL ESM without LIF addition were transferred onto the lid of a 10-cm petri dish and cultured as a hanging drop for around 7 days until EB was formed. PBS was added in the petri dish to prevent desiccation. At the end of the hanging drop culture, EBs were collected and cultured in a petri dish containing ESM without LIF addition for phase-contrast observations. To induce neuronal differentiation, D122-iPS-1C cells at 2×103 per well of a six-well plate in ESM without LIF were treated with 10−6 M retinoic acid in DMSO for 4 days. Changes in cell morphology were observed by phase-contrast microscopy, whereas immunofluorescence was used to detect the expression of neural stem cell markers.
Statistical analysis
Statistical analysis was performed by one-way analysis of variance using Prism 5 software (GraphPad). Differences with a p-value<0.05 were considered statistically significant.
Results
Generation of iPSCs from mouse Lewis lung carcinoma D122
To investigate whether mouse cancer cells can be reprogrammed into iPSCs for the purpose of generating a mouse cancer vaccine model, the mouse Lewis lung carcinoma cell line D122 was chosen because of its low immunogenicity and high metastatic property. 15 D122 cells containing a Nanog-GFP promoter reporter were reprogrammed by nucleofection with a linearized plasmid, pCAG2LMKOSimO, encoding all of the 4 Yamanaka factors, Oct3/4, Sox2, Klf4, and c-Myc, in a single transcript, and a selectable marker for neomycin resistance. 12 The transfection efficiency of the nucleofection protocol was >80%. The transfected cells were cultured according to the optimized scheme shown in Figure 1A. By day 20 after transfection, iPSC-like colonies with green fluorescence appeared (Fig. 1B–F). The frequency of forming green fluorescent colonies was between 0.005% and 0.007%. More than 200 of these colonies were then manually picked and plated on gelatin-coated plates in ESGRO medium for expansion.
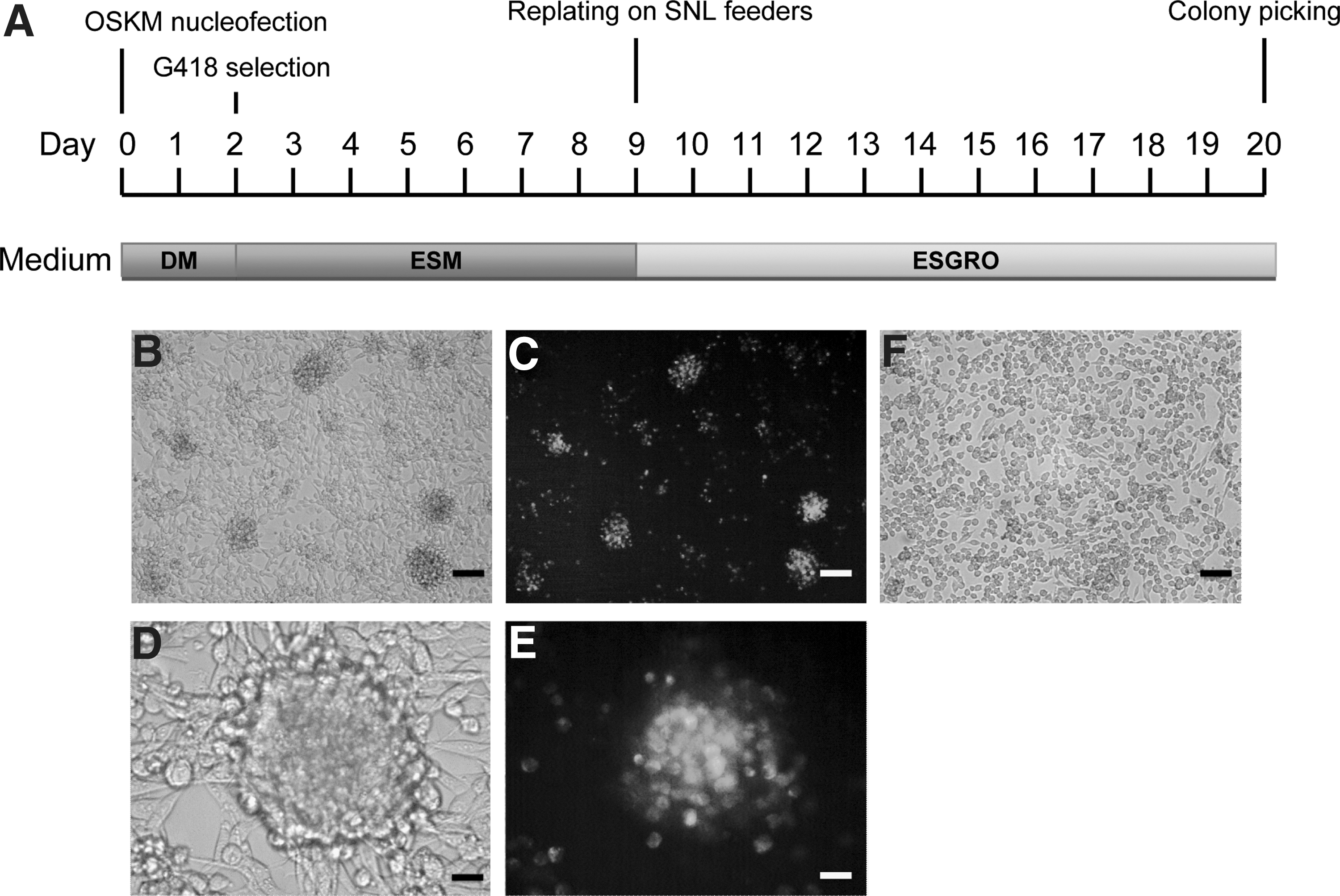
Generation of iPSCs from mouse Lewis lung carcinoma D122.
Characterization of the iPSCs reprogrammed from mouse Lewis lung carcinoma D122
Here, we describe two colonies, D122-iPS-1C & D122-iPS-1G. Both colonies maintained green fluorescence of Nanog-GFP for more than 30 passages in ESGRO medium. However, D122-iPS-1G, despite maintaining a high expression level of Nanog, displayed a substantially different expression profile of pluripotency genes compared with that in the mouse ES cells, CCB (Supplementary Fig. S1; Supplementary Data are available online at
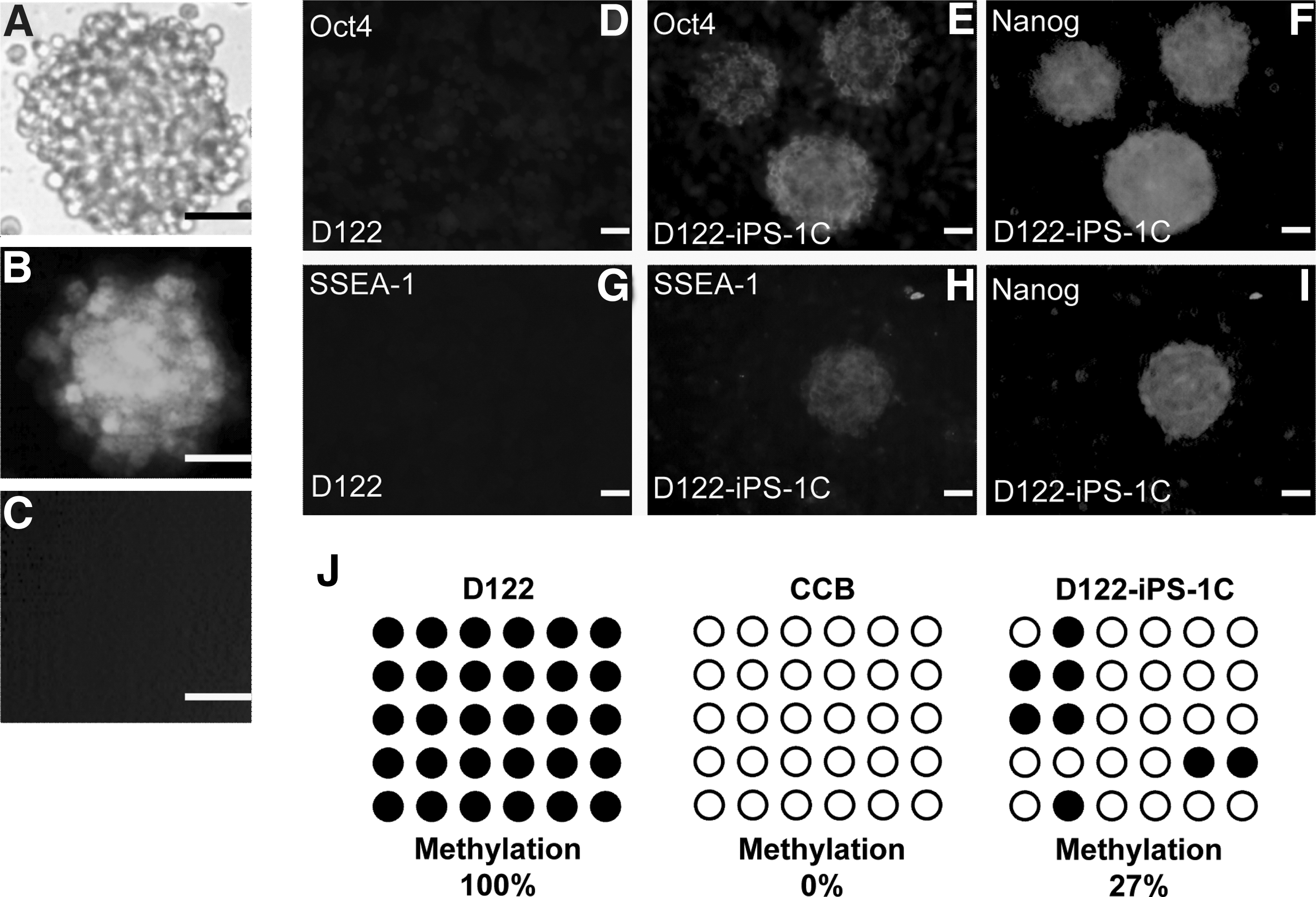
Phenotypic and epigenetic characterization of the reprogrammed D122.
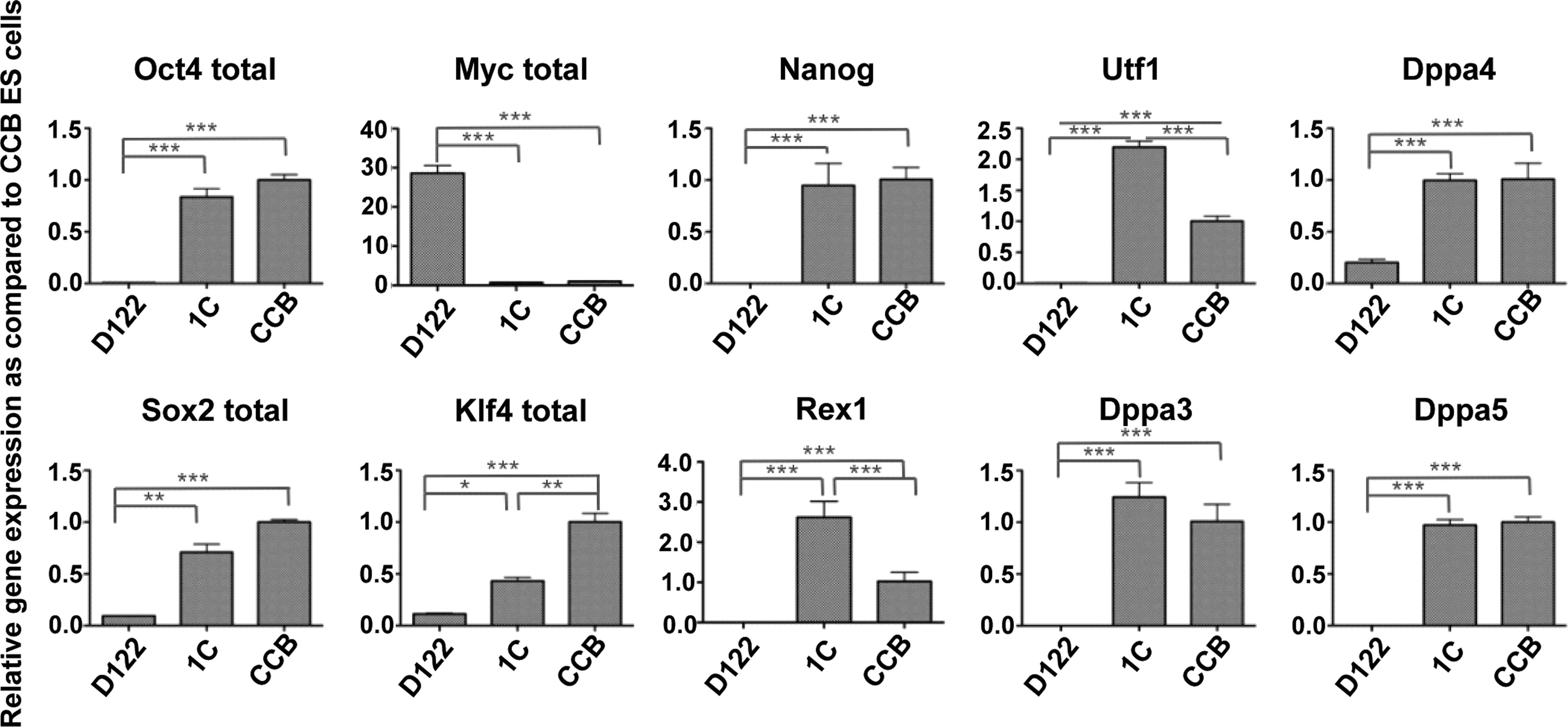
Expression profile of pluripotency marker genes of the reprogrammed D122, D122-iPS-1C (1C), in comparison with that of CCB and parental D122. Shown are the expression levels relative to those of CCB, quantified by quantitative real-time PCR using mouse TBP gene expression for normalization. Error bars represent the standard deviation of triplicate experiments. *p<0.05; **p<0.01; ***p<0.001.
Differentiation potential of the D122-iPS-1C
To determine the differentiation potential of D122-iPS-1C, EB-like clusters of these cells were collected from hanging-drop cultures and cultured on gelatin-coated plates for 1 day. Cells of neuronal, epithelial, and cobblestone-like morphologies appeared (Fig. 4A–D). To test whether D122-iPS-1C can be directed to differentiate into a specific cell lineage, we chose a simple directed differentiation model; that is, the use of retinoic acid for the induction of neuronal differentiation. After 4 days of retinoic acid treatment, a substantial fraction of these cells was found in neuronal-like morphology (Fig. 4E–G). Immunofluorescence for neural stem cell markers, PAX6 and Nestin, confirmed that 80% of these cells were indeed positive for both of these markers (Fig. 4K, O).
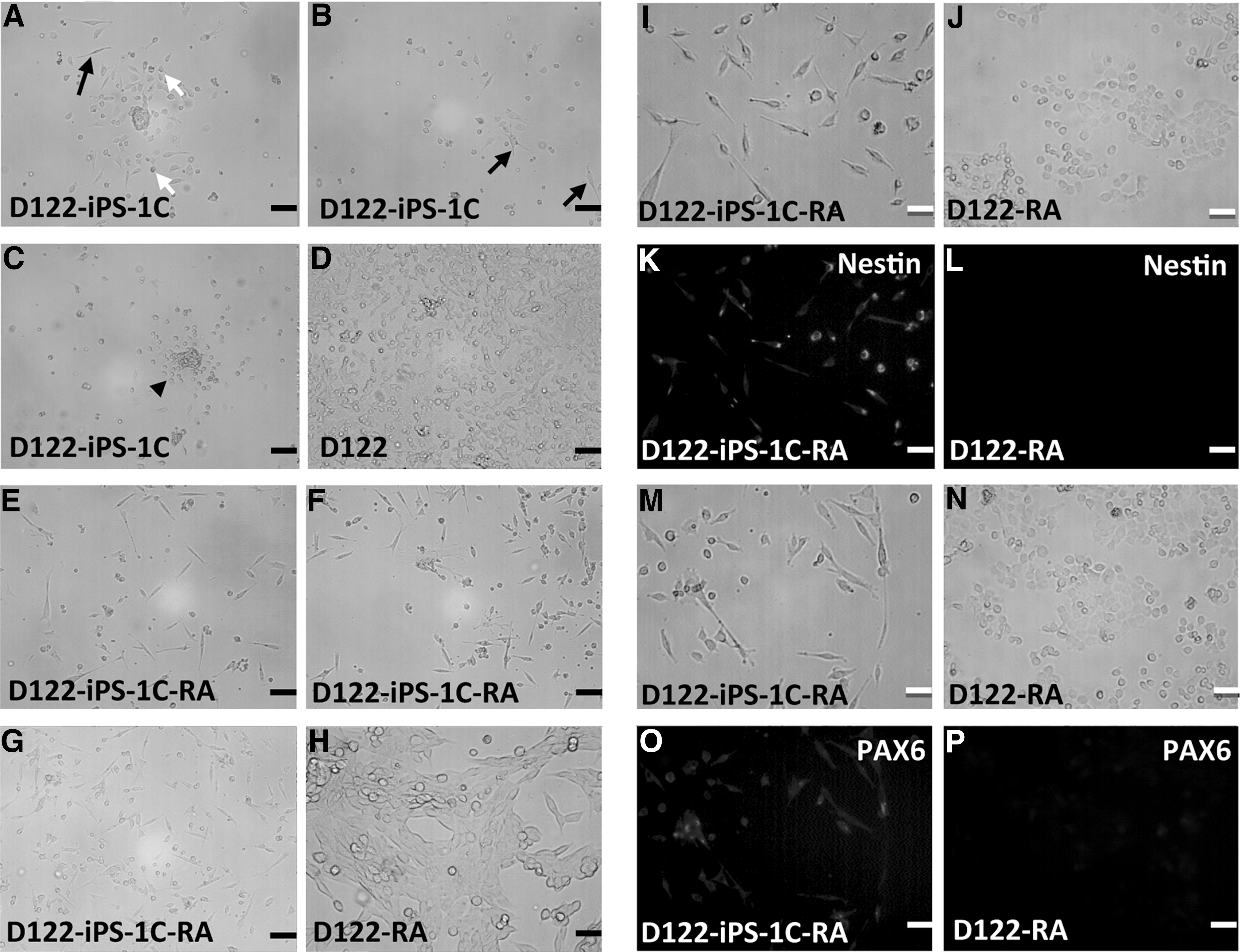
Differentiation potential of the reprogrammed D122.
Discussion
As the first step for generating mouse cancer vaccines from cancer cells via the iPSC technology, it is necessary to determine whether mouse cancer cells are amenable to reprogramming into iPSCs or iPSC-like cells that are capable of directed differentiation into a specific cell lineage. In this work, we chose a low immunogenic and a high metastatic mouse Lewis lung carcinoma cell line D122 for reprogramming, as the immunogenicity and therapeutic effectiveness of the dendritic cells derived from this cancer cell line can be unambiguously determined. Besides, mouse D122 tumor has been an established model used in immunotherapy studies. 16,17 Since dendritic cells obtained from reprogrammed cancer cells harbor the same cancer genome of the latter, these highly immunogenic cells expressing tumor antigens of the cancer cells are the best sentinel for antitumor immunosurveillance. Although our aim has been to generate dendritic cells from the reprogrammed D122, we chose neuronal lineage as the target cell type for directed differentiation at this stage because of the simplicity and short duration of the procedure compared with that required for the generation of dendritic cells. 6 –8 We wished to determine first whether the reprogrammed D122 cells had the sufficient stem cell-like property that could enable them to be differentiated into a somatic cell type. Here, we report that D122 reprogrammed by the four Yamanaka factors displayed typical characteristics of iPSCs. These included the expression of ES cell markers Nanog, Oct4, SSEA-1, and a host of other pluripotency genes at levels comparable to those in mouse ES cells, demethylation of Nanog promoter, formation of EBs, and retinoic acid-induced differentiation into neuronal cell lineage. These data together indicate that the mouse cancer cell line D122 can be reprogrammed to the stage of stem cellness which is sufficient for differentiation into other cell types. This warrants the undertaking of directing differentiation of the reprogrammed D122 into dendritic cells to be tested as an autologous cancer vaccine in the mouse D122 tumor model.
Cancer cells are not likely to be reprogrammed into such a pluripotent state that is completely indistinguishable from that of the germline-competent ES cells because of the numerous genomic mutations associated with malignancy. An earlier experiment conducted on ES cells derived from melanoma nuclei reprogrammed by somatic nuclear transfer (SNT) failed to generate germline offspring from chimeras. 18 Transfer of medulloblastoma-derived blastocysts generated by SNT into pseudo-pregnant females also failed to generate viable embryos beyond E8.5. 19 Nevertheless, these studies showed that cancer cell nuclei could be reprogrammed by the oocyte cytoplasm to become sufficiently pluripotent to support cellular differentiation into cell types of all three germ layers, give rise to early embryos, and contribute to most cell lineages in viable chimeras. 18,19 Recently, human cancer cells and EBV-transformed B cells reprogrammed by iPSC technology were shown to be able to generate typical teratomas containing derivatives of all three embryonic germ layers, and specific cell types on directed differentiation. 9,10,20 Other evidence shows that partially reprogrammed germline-incompetent iPSCs are sufficiently pluripotent to differentiate into multiple somatic cell lineages. The earliest report on iPSC generation demonstrated that the fbx15-selected iPSCs, while being only partially reprogrammed as indicated by the Nanog and Oct 4 promoters retaining substantial methylation, were able to differentiate into cell types of all three germ layers, and to contribute to early mouse embryonic development. 1 Consistently, partially reprogrammed human fibroblasts showing no or only some Nanog expression were shown to be capable of mesoderm differentiation in teratomas. 21 Since the hematopoietic system, from which dendritic cells arise, originates from the mesoderm, the observation just cited particularly supports the feasibility of generating dendritic cells from reprogrammed cancer cells. In our present study, as indicated by the extensive demethylation of the Nanog promoter, the D122-derived iPS-like cells were better reprogrammed than the fbx15-selected iPSCs and partially reprogrammed human fibroblasts just mentioned.
We are aware of the possibility that severe chromosomal abnormalities in some cancer cells may render the cells unable to be reprogrammed or differentiate into the required specific cell types. Thus, this may present a limitation for the general applicability of using iPSC technology to generate autologous cancer vaccines. Nevertheless, we demonstrated that it is possible to reprogram mouse cancer cells to such a state of pluripotency that subsequent differentiation into a specific cell lineage is possible. It is, thus, feasible to proceed to the generation of dendritic cells from reprogrammed mouse cancer cells to determine the effectiveness of this novel approach of cancer immunotherapy in mouse tumor models.
Footnotes
Acknowledgments
The authors gratefully acknowledge Ms. Peggy Hoi-Ying Fung for clerical help in preparing this article, and Mr. Chun-Hung Ma for technical assistance. This work was supported by a Direct Grant for Research Reference number 2010.1.024. The anti-Nestin monoclonal antibody (Rat-401) and anti-PAX6 monoclonal antibody, developed by Hochfield, S and Kawakami, A, respectively, were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
Disclosure Statement
There are no existing financial conflicts.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.