
Abstract
Early and specific tumor detection and also therapy selection and response evaluation are some challenges of personalized medicine. This calls for high sensitive and specific molecular imaging such as positron emission tomography (PET). The use of peptides for PET molecular imaging has undeniable advantages: possibility of targeting through peptide-receptor interaction, small size and low-molecular weight conferring good penetration in the tissue or at cellular level, low toxicity, no antigenicity, and possibility of wide choice for radiolabeling. Among β+-emitter radioelements, Gallium-68 is a very attractive positron-emitter compared with carbon-11 or fluorine-18 taking into account its easy production via a 68Ge/68Ga generator and well established radiochemistry. Gallium-68 chemistry is based on well-defined coordination complexes with macrocycle or chelates having strong binding properties, particularly suitable for linking peptides that allow resistance to in vivo transchelation of the metal ion. Understanding specific and nonspecific molecular mechanisms involved in oncogenesis is one major key to develop new molecular imaging tools. The present review focuses on peptide signaling involved in different oncogenic pathways. This peptide signalization might be common for tumoral and non-tumoral processes or could be specific of an oncological process. This review describes gallium chemistry and different 68Ga-radiolabeled peptides already in use or under development aiming at developing molecular PET imaging of different oncological processes.
Introduction
The anticipated success of personalized medicine depend on molecular-targeted drug having a linked diagnostic test designed to precisely determine whether a patient will benefit from the specific treatment or to monitor targeted therapy as a way to determine ongoing efficacy. Consequently, early and pathological-specific process detection of tumors in humans is one of the challenges of theranostics (i.e. the merging of drug therapy and diagnostics to advance personalized medicine). In this context, molecular imaging plays a leading role in exploring, in a given patient, pathogenesis-related specific biochemical dysfunctions. Among molecular imaging techniques, nuclear methods are by far the most sensitive. Apart from localization and quantification, the most important advantage of such molecular imaging is the opportunity it provides to investigate the dynamics of pathogenesis-specific molecular process in vivo. Due to its high sensitivity for detection of small amount of radioactivity emitted by the radiopharmaceuticals of known chemistry, and the possibility of quantification, Positron Emission Tomography (PET) is now widely utilized. PET has been used as a research tool for more than two decades, and it affords researchers the ability to conduct both functional and molecular imaging on biological and biochemical processes in vivo. In functional imaging, biological parameters such as metabolic rate and perfusion that can be altered by diseases or treatment are monitored. In molecular imaging, PET can be used to examine and quantify cellular events such as cell trafficking, receptor binding, and gene expression. Therefore, PET is an important modality to elucidate mechanisms associated with diseases and drug interactions. Today, PET is used as a powerful nuclear medicine technique for diagnosis and therapy follow-up of many tumors, but it is not pathogenesis-specific because the most currently used radiotracer is [18F]-2-fluoro-deoxy-D-glucose ([18F]-FDG) that only reveals in vivo metabolic activity. 1
In the last decade, several new β+ isotopes have been produced promoting PET development (see Table 1). 2 –5 The choice of the PET radionuclide is imposed by its physical half-life, positron branching, production mode, chemistry involved in radiolabeling, and potential effects on the intrinsic spatial PET resolution. For example, radionuclides such as 66Ga or 86Y with high β+ maximum energy emit positrons with high range in the matter, which considerably lower spatial resolution particularly important during preclinical studies using μPET imaging system. They have to be carefully manipulated.
Most β+-emitters are still dependent on an on-site cyclotron. Until nowadays, the two major positron-emitters mainly used for PET molecular imaging are 11C and 18F. They present some limitations in radiopharmaceutical development, namely
• 11C: the ubiquitous presence of carbon in any organic molecule makes 11C an attractive radioisotope for labeling biomolecules because it does not change the chemical structure and the biological properties of the original molecule. For instance, 11C-labeled methionine is derived from its precursor homocysteine thiolactone rendering methyl group in the system. 6 Several other 11C-labeled compounds that sustain their biological integrity like [11C]-Choline, [11C]-Acetate are routinely in use. 7 Almost all syntheses involving 11C begins with [11C]CO2 and [11C]CH4 as primary products. The most widely used method for introducing 11C into a biomolecule is an alkylation reaction, using functional groups such as [11C]CH3I or [11C]CH3OTf, which are derived from the primary products. However, the syntheses with 11C present several features: limited number of labeled precursors, submicromolar amounts of the starting materials, and necessity of development of rapid synthetic procedures (multistep syntheses are not generally used for the synthesis of 11C-labeled molecules) to incorporate the 11C radioisotope into the biomolecule. Due to these reasons and its relatively short half-life (20.37 minutes), and the necessity of a very closed biomedical cyclotron to PET camera, 11C has been less developed to clinical applications as opposed to its 18F counterpart.
• 18F could act as a bioisosteric replacement for a hydrogen atom in a molecule and may imitate a hydroxyl group. Radiochemistry with 18F implies special challenges and direct incorporation (i.e. 18F is introduced in one step using [18F]F− ion as reagent for nucleophilic reactions or [18F]F2 as reagent for electrophilic reactions) of 18F at high specific radioactivity into peptides or proteins is not possible due to the harsh reaction conditions needed during labeling reaction (high temperature and high alkalinity), which may decompose or denature the peptide of interest. Nowadays, direct labeling is used for the routine synthesis of [18F]-FDG 8 and [18F]-FDOPA. 9
To overcome the risk of denaturation, radiofluorination procedure can be performed using fluorinated prosthetic groups that will then react with functional groups of peptides (e.g. carboxylic acid and amine or sulfhydryl-reactive bioconjugate) under mild conditions. 10,11 This strategy requires a multistep approach and several reactions (synthesis and purification) that lengthen the time required for the radiosynthesis are needed. Thereby, as for 11C, 18F-labeling procedures are performed in an in-site biomedical cyclotron. [18F]-SFB (N-succinimidyl,4-[18F]fluorobenzoate) is an example of prosthetic groups used for indirect 18F-labeling that reacts with lysine residue of proteins. 12
Another option to support hospitals with relevant PET radionuclides is using radionuclide generators.
13
–17
Generators-based radionuclides would allow easier availability and more flexibility in use. The decay of a long-lived parent nuclide to a short-lived PET daughter radionuclide provides an inexpensive and convenient alternative. As a consequence, 68Ga has emerged with high potential in clinical PET molecular imaging. Several advantages over 11C and 18F, for the development of radiopharmaceuticals are as follows: • A production much more easier than 11C and 18F because it comes from the decay of 68Ge in 68Ga in an in-house 68Ge/68Ga generator; 68Ga is thus available “on demand” after elution of the system, similar to the production of 99mTc, unlike 11C and 18F are cyclotron-produced isotopes. The usefulness of imaging based on 68Ge/68Ga systems are supported by a large number of studies on 68Ga-labeled molecules.
18
–26
So, 68Ge/68Ga generator might become in future what 99Mo/99mTc generator is today for nuclear medicine. • Because of its advantageous half-life (67.71 minutes), 68Ga is a valuable metallic radionuclide in PET suitable for the pharmacokinetics of many peptides. • Finally, 68Ga has to be coordinated by a macrocycle to be introduced in a targeted peptide. This chelate allows formation of stable complexes with various trivalent radiometals for imaging or targeted radionuclide-therapy applications paving the way for multimodal imaging agents including PET-MRI.
However, the introduction of this macrocycle may alter the pharmacological and/or pharmacokinetics properties of the biomolecule. Reubi et al. have shown that addition of a DOTA chelator to a pan-somatostatin analog, named 406-040-15, triggers a switch at somatostatin receptor subtype 3 (sst3) receptor from an antagonist to an agonist profile. 27 So, macrocycle linked bioconjugate could exhibit different pharmacological properties compared to the precursor and has to be biologically validated.
In the present article, we will first discuss about gallium chemistry; isotopes and molecular imaging agents based on gallium use and then describe the different processes, specific and nonspecific, involved in carcinogenesis, focusing on the ability to target processes with peptides radiolabeled with 68Ga under development.
Gallium
Gallium chemistry
Gallium is a strong Lewis acid because of its high charge density and small ionic radius (62 pm) and could form thermodynamically stable complexes with strong Lewis bases. 28,29 In aqueous solutions, gallium is stable only as a trivalent cation Ga3+ under acidic conditions. (The low-valent oxidation state (+I) is not of significance in aqueous media and has no relevance in the radiolabeling of radiopharmaceutics). pKa value of Ga3+ ion in its hydrated form is 2.6. 28 Between pH 3 and 9, insoluble gallium [Ga(OH)3] is the predominant species, whereas at pH >9.6 soluble gallate ion [Ga(OH)4]- formation begins.
The Ga3+ cation has a d10 electron configuration and accepts different coordination numbers (usually 4–6), while not displaying preferences for particular coordination polyhedron. Complexation of Ga3+ ion is mainly based on ligands containing oxygen, nitrogen, and sulfur donor atoms.
Gallium isotopes
Naturally occurring gallium consists of two isotopes, 69Ga and 71Ga (respectively 60.1% and 39.9% natural abundance). Three radioisotopes of gallium are available for labeling of peptides and small molecules. Two of these, 66Ga (T1/2=9.49 hours) and 68Ga (T1/2=67.71 minutes), decay by positron-emission and are therefore suitable for PET imaging, 67Ga (T1/2=78.3 hours) decays by gamma-emission and is used for SPECT imaging. 68Ga is a short-lived positron-emitter available through the 68Ge/68Ga radionuclide generator system. The long-lived 68Ge (T1/2=270.9 days) is accelerator-produced by the 69Ga (p,2n) 68Ge nuclear reaction and decays by pure orbital electron capture and produces a short-lived 68Ga, which is a positron-emitter (high β+-branching of 89%, Eβ + max=1899 keV). 30 Commercially available generators contain 68Ga absorbed onto a solid TiO2 chromatographic support, generators based on a SnO2 solid support are also available. Recently, other kinds of supports have been proposed. Chakravarty et al. developed a nano-zirconia based 68Ge/68Ga generator 31 and Nakayama et al. built a column containing an N-methylglucamine group linked to styrene-divinylbenzene copolymer beads, which exhibits high affinity to 68Ge and low affinity to 68Ga allowing elution of 68Ga from the system. 32
Gallium-based molecular imaging agents
The design of most molecular imaging agents is based on the use of bifunctional chelating agents, that is, a “platform” able to coordinate the radiometal on one side and to conjugate biomolecules through an appropriate functional group on the other side.
Like other metallic radionuclides ions (Y3+, In3+, Cu2+, Zr4+, Sc3+), gallium cannot be incorporated into radiopharmaceuticals vectors by covalent bonding, but has to be complexed with a chelate using well established coordination chemistry. Two main conditions are needed to develop suitable gallium-based radiopharmaceuticals; on the one hand, stability to hydrolysis to prevent formation of insoluble and soluble gallium hydroxides and on the other hand, 68Ga-complexes have to be more stable than 68Ga-transferrin complex to avoid transchelation because transferrin displays two iron binding sites with high affinity for this metal ion. (Ga3+ and Fe3+ have similar oxidation state and ionic radii, respectively 62 pm and 65 pm, and the formation constant of Ga-transferin complex, logK1 , is 20.3). 28,33 –36
For in vivo applications, kinetic inertness can be more important than thermodynamic stability and strong ligand coordination is always required to achieve sufficient stability.
Generally, acyclic chelator complexes are less kinetically inert than macrocycle complexes of comparable stability. Most chelating agents that form highly stable complexes with Ga3+are hexadentate, to sequester Ga3+ up to its maximum coordinate number of six, and contain carboxyl, amino, or thiol groups. Polyaminocarboxylates macrocycles such as 1,4,7,10-tetraazacyclododecane-N, N′, N′′, N′′′-tetraacetic acid (DOTA), or 1,4,7-triazacyclononane-N, N′, N′′-triacetic acid (NOTA) and its NCS or N-hydroxysuccinimide derivates, for example, (DOTA-NCS or DOTA-NHS) respond to these characteristics and show excellent in vivo stability (Fig. 1). Other tetra-or pentadentate ligands have also been described. 33
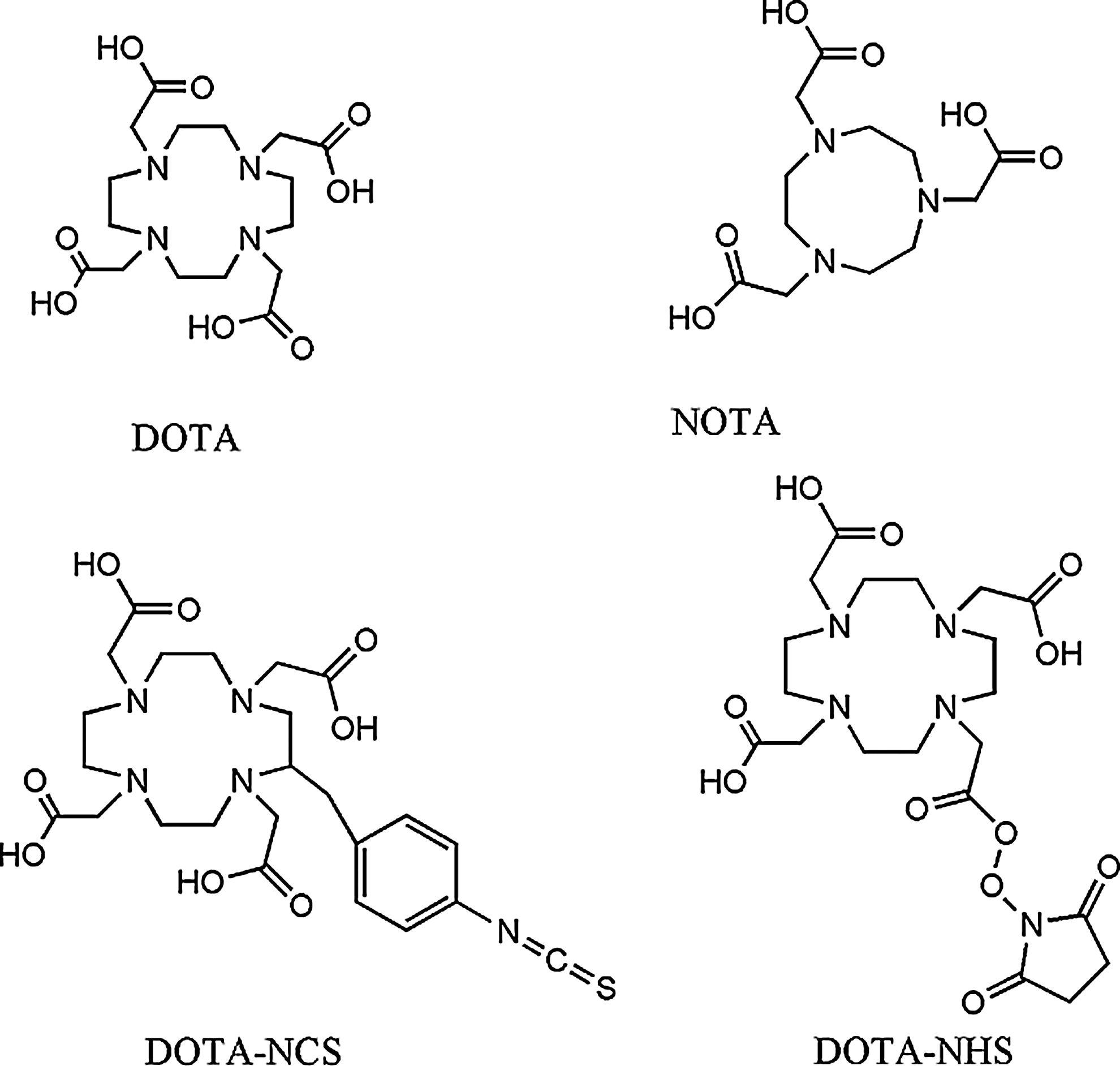
Structure of 1,4,7,10-tetraazacyclododecane-N, N′, N′′, N′′′-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-N, N′, N′′-triacetic acid (NOTA), and some derivates.
DOTA is potentially octadentate while NOTA is hexadentate. So, DOTA can more than saturate the usual six-coordination sphere of Ga3+. A study led by Viola et al. was carried out to better understand Ga-DOTA complex structure and geometry (complex was formed by reaction between gallium chloride and DOTA macrocycle in aqueous solution under acidic condition (pH value 5), with regard to reaction stoichiometry). This consists of a N4O2 donor set involving an equatorial plane composed of two nitrogens from the ring and two oxygens from the carboxylate group. The plane formed is slightly distorted. The third carboxylate group is deprotonated and does not coordinate Ga3+, while the fourth carboxylate group is conjugated to an amino acid group belonging to the targeted peptide) and the Ga-DOTA geometry is a pseudo-octahedral structure involving distortion of DOTA-cavity resulting in a cis-coordinated complex (Fig. 2). 37,38 Thus, in the molecular design of bifunctional chelators, the use of a single pendant carboxylate group of DOTA for conjugation to the targeted vector does not affect its gallium binding capacity because only six out of N4O4 donors are used in bonding Ga3+ ion. In the case of NOTA, this is not true because the three carboxylate groups are involved in Ga3+ coordination.

Grafting a DOTA-radiometal complex on a biomolecule. Image taken from Viola et al. Reprinted with permission from reference number 2885260532558.
DOTA has a larger cavity than NOTA, which results in lower thermodynamic stability of the 68Ga3+ complex (thermodynamic stability constants for Ga-NOTA and Ga-DOTA complexes are respectively 30.98 and 21.33), 11,28 and consequently longer reaction times. This is because the ring distortion is determining reaction conditions for labeling. As DOTA needs a higher ring distortion than NOTA to label Ga3+ ion, complex formation with DOTA, requires high temperature (90°C–100°C during 10 minutes using microwave), whereas complex formation with NOTA could be performed at room temperature. Moreover, the compactness of NOTA leads to high stability and selectivity for Ga3+. 39 The hole-size effects influence both thermodynamics and kinetics of macrocycle complexes. This is because a chelator in its minimum energy metal-binding conformation will be optimized for a particular size of a metal ion, and when other metallic radionuclides are bound the macrocycle conformational energy will increase with a resultant decrease in complex stability. 40
Although NOTA is particularly suitable for complexing 68Ga3+ ion, we believe that the use of DOTA macrocycle (and its derivates) is of great interest because larger metallic radionuclides such as 213Bi, 86/90Y, 89Zr, or 44/47Sc could be coordinated paving the way to imaging or targeted radionuclide-therapy applications. Radiolabeled DOTA-TOC with 68Ga, 111In, 177Lu, 90Y, or 44Sc and DOTA-derivated bombesin analog labeled with 213Bi or 86Y have been investigated. 41 –46
Metallation reactions with DOTA are performed using a buffer system such as sodium acetate, HEPES, phosphate, or ammonium acetate to adjust the pH of the 68Ge/68Ga generator eluate closed to pH value 4. This condition is a prerequisite, as discussed in Gallium chemistry paragraph, for ionizing 68Ga and complexing DOTA-based biomolecules with this metallic radionuclide because at pH value >4, formation of colloidal hydroxide [Ga(OH)3]n begins. Although this does not generally inhibit complex formation, radiolabeling is nevertheless substantially hampered due to formation of insoluble colloids and their adhesion to the surface of the reaction vessel. At pH values above 9, a water-soluble hydroxo complex [Ga(OH)4]-, is formed. As ligand exchange with the tetrahydroxo complex is a much slower process than complexation of ionic Ga3+, metallation is achieved best at a maximum pH value 4. Metal cation contaminants (Fe3+, Zn2+, Ge4+, Ti4+, etc.) coming from the 68Ge/68Ga generator eluate, glassware, or from other reagents (HCl, acetate buffer etc.) could hamper achieving a satisfactory 68Ga labeling yield and specific activity (stability constant of Fe-DOTA complex is 29.4). 47 To avoid high radiation doses, the radiolabeling procedure is mostly performed in synthesis modules housed in lead shielded hot cells.
Introducing macrocyles into peptides is a challenge and need the development of efficient methods. Because of the similarity between DOTA and NOTA macrocycles we will only discuss the two main strategies for introducing DOTA macrocycle in biomolecules (Fig. 3). 39

(Up) Pathways of synthesis using tris-tert-butyl-DOTA and tris allyl-DOTA. (Down) Reaction using DOTA-OSSU. Image taken from Tanaka and Fukase. Reproduced by permission of The Royal Society of Chemistry.
One consists of the use of coupling agents: tert-butyl and allyl protecting groups are carboxylic acid functional groups of side chains that react with the desired amine groups of the peptide. The synthesis is performed on a solid phase and includes two stages: the first is the reaction of the protected DOTA with the amine of the peptide by coupling agents, then the deprotection of acid functions with trifluoroacetic acid (TFA) for tris-tert-butyl-DOTA and Pd(PPh3)4, morpholine and TFA for tris-allyl-DOTA.
The other strategy uses activated ester of DOTA. This conjugation step can be carried out by reacting a molar excess of the activated tri-tert-butyl ester-derivatized chelator with a designated free amino group of a desirable peptide, in which all other reactive amines are protected, in the presence of a coupling agent. For instance, DOTA-O-sulfosuccinimidyl ester (DOTA-OSSU).
DOTA-OSSu prepared in situ from DOTA, EDC (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide), and N-hydroxysulfosuccinimide is intended to directly react with the amine group of the biomolecule to give DOTA conjugates.
Combining peptides advantages (i.e. possibility of targeting through peptide-receptor interaction, small size and low-molecular weight conferring good penetration in the tissue or at cellular level, low toxicity, and no antigenicity), macrocycle chemistry richness and 68Ga physical properties and its availability through a 68Ge/68Ga generator, we are convinced that 68Ga-radiolabeled peptide-based PET molecular imaging has a promising future.
Mechanisms Involved in Tumoral Processes and Targeting with 68Ga-Radiolabeled Peptides
Several mechanisms that are important in the carcinogenic process are common to tumoral and nontumoral processes but their dysregulation is often prominent in tumors. They include, but are not limited to, adhesion phenomenon, proliferation process, apoptosis, tumor invasion, and inflammation. For more clarity we will only discuss their implication in oncology.
Adhesion phenomenon
Adhesion molecules are both anchorage and signalization molecules. They allow the maintenance of cellular architecture and actively participate in the cellular homeostasis. Their dysregulation during oncogenesis is common. Among these adhesion molecules, integrins ligands have been mainly studied for oncogenesis imaging. Integrins are heterodimeric transmembrane glycoproteins constituted by an α chain and a β chain.
In human, 18 different α and 8 different β subunits are described, which can combine into 24 different integrins (Fig. 4). 29
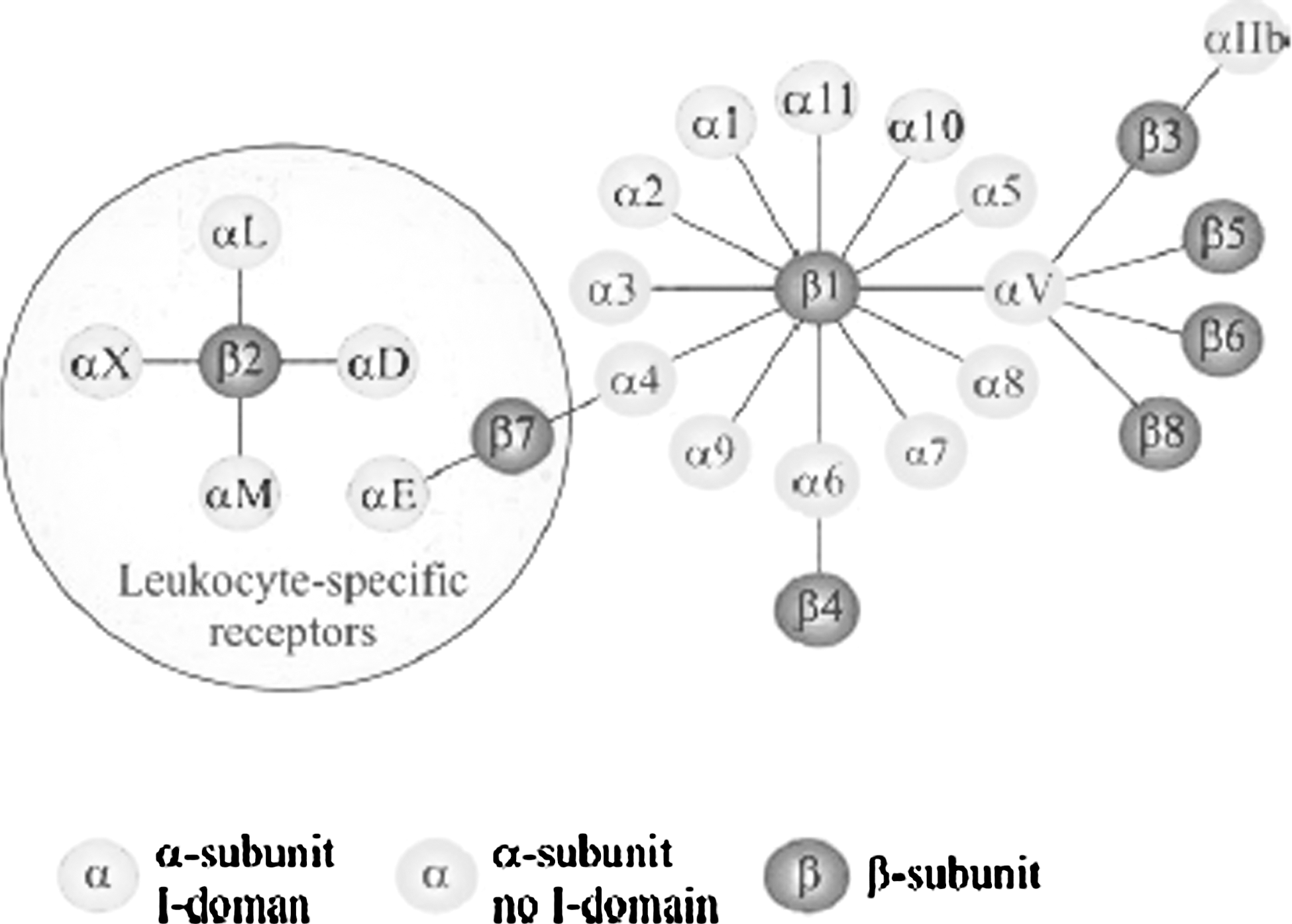
The integrin superfamily. Image taken from Niu and Chen.
Overexpression of surface adhesion molecules such as integrins and loss of some cell adhesion molecules are responsible for the acquisition of invasion, metastasis, and neoangiogenesis properties. 48 For example, integrin αvβ3 expression is stimulated by angiogenic factors such as β fibroblast growth factor or tumor necrosis factor α (TNFα) and promotes angiogenesis. Integrin αvβ5 is another integrin implicated in angiogenesis. 29
It was found that the amino acid sequence Arg-Gly-Asp (RGD) is an important binding epitope of several extracellular matrix proteins (collagen, fibrinogen, and vitronectin).
Several 68Ga-radiolabeled peptides selectively binding to integrin αvβ3 are under investigation. We can mention [68Ga]Ga-NOTA-G3-RGD2 49 synthesized, radiolabeled, and evaluated by Liu et al. and [68Ga]-NODAGA-RGD by Knetsch et al. 50 They respectively have been evaluated on human glioblastoma U87MG (αvβ3 positive) and on human melanoma M21 (αvβ3 positive). Integrin binding affinity of NOTA-G3-RGD2 by competitive displacement of [125I]-echistatin bound to U87MG cells shows an IC50 of 66.4 nM, revealing a good affinity. In vivo studies, performed 1 hour after injection, show radiotracer accumulation into kidneys and urinary excretion.
Proliferation process and growth factors
Mitotic signals are, in healthy cells, processes tightly regulated to control/regulate cell proliferation.
One of the major molecules found in the transmission of these signals is the human Epidermal Growth Factor (hEGF), which binds to its receptor (HER). HER family is composed of four receptors: HER1/EGFR, HER2, HER3, and HER4.
HER has an extracellular domain to which hEGF binds and a transmembrane portion and an intracellular ATP-binding domain with tyrosine kinase activity responsible to signaling pathways. Each HER molecule exists in a monomer form when inactive, converting into dimer when becoming active. Different signal transduction pathways are involved after receptor activation such as Mitogen Activated Protein Kinase (MAPK) or Akt. 51 hEGF and other growth factors (VEGF, PDGFetc.) and their tyrosine kinase receptors play an important role in cell proliferation, apoptosis, adhesion, migration, and cell differentiation. Today, there are strong arguments in favor of their role in oncogenesis. 51,52 Several oncogenic alterations have been identified, both in the extracellular domain and in intracellular proteins involved in signal transduction. For example, EGFR vIII neither binds to ligands nor dimerizes because of a mutant tyrosine kinase domain that is constitutively activated. As for HER2, it is overexpressed in many cancers: breast, gastric, esophageal, colorectal, lung, prostate, bladder, pancreatic, and ovary. Treatment with trastuzumab, an antibody directed to wild HER2 has improved survival of patients with HER2 overexpressing breast tumors. It is now well known that HER2 overexpression is associated with adverse prognosis in breast cancer. 52
Tolmachev et al. have developed a HER2-binding affibody labeled with 68Ga: [68Ga]-DOTA-Zher2: 342-pep2. It is an antibody directed to HER2. In vitro studies were performed on SKOV3 cells (human ovarian cancer cells with high level of HER2 expression). Kd is 78 pM, which is a very good result revealing a strong affinity. Biodistribution studies were performed on mice bearing SKOV3 xenografts. These studies show good tumor specificity and rapid renal clearance. 52 A study on 3 patients with breast cancer was performed by Baum et al. (Fig. 5). 53 HER2 overexpressing metastatic lesions were clearly visualized.

HER2 expressing metastasis (arrows). Image taken from Baum et al.
Apoptosis
In the body, and in different organs and tissues, the total number of cells is the result of tightly regulated balance between mitosis and apoptosis. An imbalance between these two functions can promote carcinogenesis. 54 The process of apoptosis, involves two pathways called intrinsic and extrinsic. 55,56 Mitochondrial or intrinsic pathway is particularly activated as result of DNA damage or cellular damage. Then, there is a mitochondrial membrane pore opening releasing cytochrome c, leading to in caspase 3 activation, apoptosis effector, via formation of an apoptosome. The extrinsic pathway is activated by TNF, FasL binding on cell membrane receptors. Adapter proteins like FADD are brought into play in addition to intracellular signaling pathways and activation of caspase 8. Caspase 8 can cleave the procaspase 10 into caspase 10 or activate the mitochondrial relay via Bid. After caspase 3 activation, there is a rapid redistribution and exposure of phosphatidylserine to the cell surface. Phosphatidylserine is absent from the basolateral leaflet of the membrane of healthy living cells and is located in the plasma membrane of dying cells. Annexin A5 is a protein that specifically binds to phosphatidylserine with high affinity. 57,58
Induction of apoptosis is the primary mechanism through which most chemotherapies cause tumor cell death.
Follow-up of response to cancer therapy is crucial for an optimal treatment strategy to reduce the dose in a responding patient or to change to another therapy type in a nonresponding patient. Molecular imaging of apoptosis offers a direct and early measurement of response to cancer therapy.
Two peptides derived from annexin A5 radiolabeled with 68Ga were developed by Bauwens et al. 59 while maintaining the binding capacity of annexin A5 to phosphatidylserine: the [68Ga]-Cys2-AnxA5 and [68Ga]-Cys165-AnxA5. Biodistribution and pharmacokinetics were evaluated before and after treatment with cyclophosphamide in mice bearing xenografts of human B lymphoblast cell line Daudi.
Biodistribution of both tracers proved to be similar with fast clearance from the blood and a high retention in the kidneys. Tumor retention before chemotherapy is low for both radiotracers but was significantly increased following chemotherapy-induced apoptosis. Derivates of annexin A5 radiolabeled with 68Ga could be helpful for early evaluation of antitumor therapy.
Tumor invasion
Matrix metalloproteinases (MMPs) are a family of zinc-dependent endopeptidases characterized by their ability to degrade extracellular matrix components. They play an important role in shaping and tissue remodeling in physiological or pathological conditions (Fig. 6). Human MMP family is composed of 23 members. For example, MMP9 is found in keratinocytes, neutrophils, and eosinophils. 60
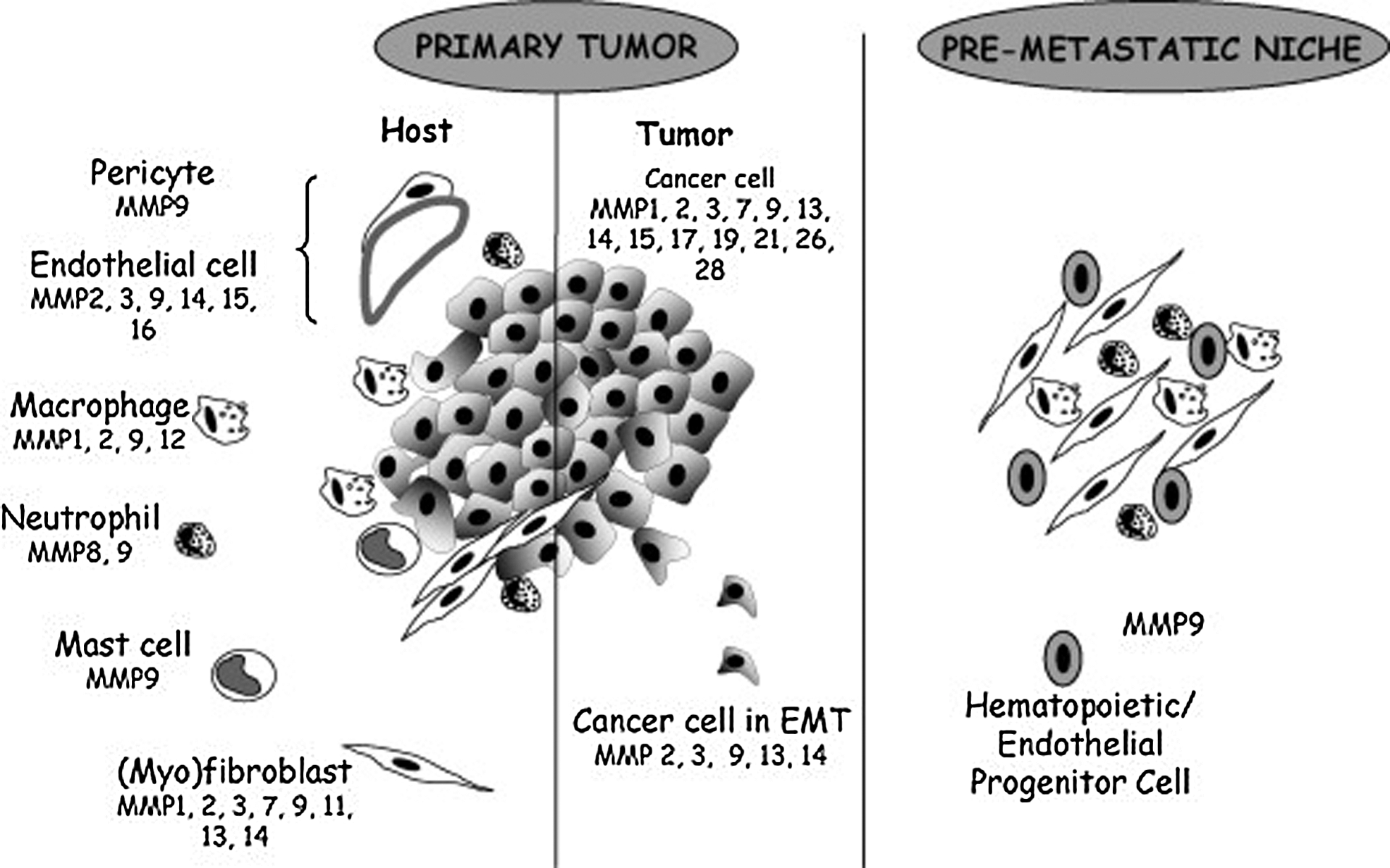
Matrix metalloproteinases expression. Image taken from Noel et al. Reprinted with permission from reference number 2884811102429.
MMPs play an important role in various aspects of cancer progression. MMPs degrade extracellular matrix proteins, thus promoting tumor cells migration into the blood and surrounding tissues, beyond the immune system. In addition, this degradation allows the release of growth factors that are sequestered. For example, MMP9 mobilizes VEGF from the matrix. MMP9 overexpression is associated with invasion and metastasis or rapid tumor progression in patients with melanoma. 61,62
Ujula et al. have synthesized and radiolabeled with 68Ga two analogs of Tumor Cell Targeting Protein (TCTP-1), a ligand of MMP9. One of them, the [68Ga]-DOTA-TCTP-1 is cyclized by a cysteine bridge.
In vitro experiments show a high hydrophilicity. In vivo stability experiments reveal a good stability in human plasma and a half-life of 2.5 hours. In vivo PET imaging after human melanoma cells C8161T/M1 xenografting in athymic rats shows uptake in the tumor (Fig. 7). 61
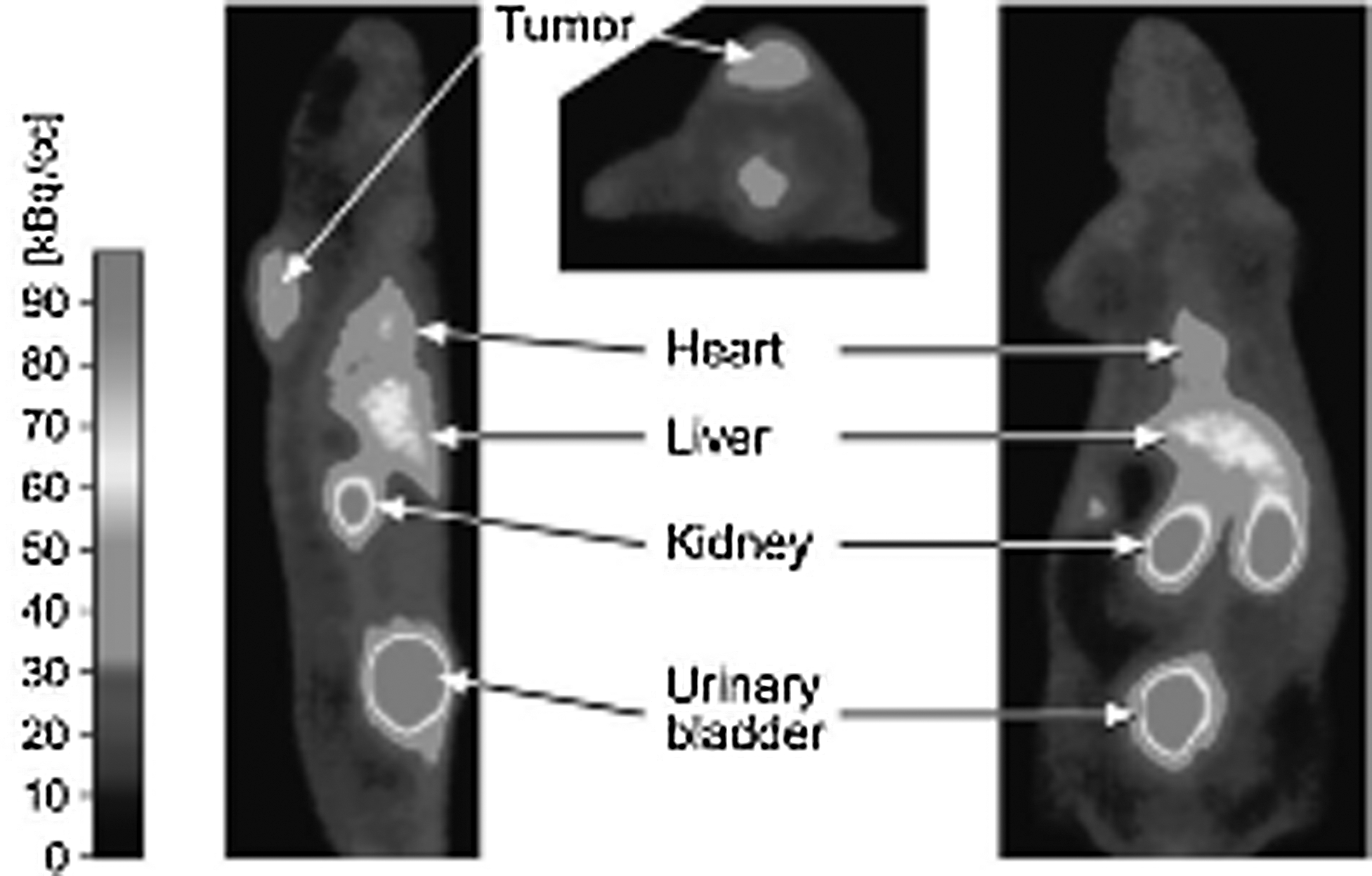
Positron emission tomography (PET) imaging. Image taken from Ujula et al. Reprinted with permission from American Chemical Society Copyright (2010).
Oncological Processes and Targeting with 68Ga-Radiolabeled “Hormone-Peptides”
In this section we describe other mechanisms involved in oncological processes represented by hormone peptides and their receptors. We will successively focus on somatostatin, bombesin, neurotensin, neuropeptide Y, “glucagon-like” peptide, “melanocyte stimulating” hormones, and “gonadotrophin releasing” hormones.
Somatostatin and neuroendocrine tumors
Somatostatin is a peptide hormone that binds with high affinity to five different subtypes of specific somatostatin receptors (sst1 to sst5). They belong to the G-Protein Coupled Receptor (GPCR) superfamily.
The majority of neuroendocrine tumors (NET), and some other tumors, such as small cell lung cancer, breast tumor, and malignant lymphoma, overexpress multiple somatostatin receptor subtypes, of which the sst2 subtype is predominantly expressed. 63,64
Somatostatin is not a good peptide to label because of its very short plasma lifetime (about 3 min). Analogs of longer half-life were synthesized such as octreotide, lanreotide, or pentetreotide. 65 Today, somatostatin receptor scintigraphy with [111In]-DTPA-pentetreotide (Octreoscan®) allows NET detection with high sensitivity and good specificity for detection of the primary tumor and secondary lesions. Yet, sensitivity of [111In]In-DTPA-pentetreotide scintigraphy is limited because a number of tumors overexpress other subtypes of somatostatin receptors and also because the rather low spatial resolution limits the ability to detect lesions with small size and lower receptor density. 66
Number of peptides have been evaluated for PET detection such as [68Ga]-DOTA-Tyr3-octreotide-Phe1 ([68Ga]-DOTA-TOC), which binds to sst2 with high affinity. Many studies have shown the superiority of [68Ga]-DOTA-TOC PET over Octreoscan SPECT in patients with NET. 67
Other octreotide derivates have been synthetized, radiolabeled with 68Ga, and evaluated in the management of NET: [68Ga]-DOTA-NaI3-octreotide ([68Ga]-DOTA-NOC) 19,68,69 and [68Ga]-DOTA-Tyr3-octreotate ([68Ga]-DOTA-TATE). 70,71
Poeppel et al. have published a study comparing the use of [68Ga]-DOTA-TOC and [68Ga]-DOTA-TATE in NET imaging and some differential uptake has been shown. 72
[68Ga]-DOTA-NOC and [68Ga]-DOTA-TATE are today clearly superior to somatostatin receptor scintigraphy for the assessment of NET.
Gastrin-releasing peptide and other bombesin-related peptides
Bombesin was discovered in the frog “Bombina bombina.” Two related peptides are present in man: gastrin-releasing peptide (GRP) and neuromedin-B. They are members of the brain-gut peptides present in the central nervous system, gastrointestinal tract, and the pulmonary tract. They regulate many biological functions including secretion of gastric acid, regulation of body temperature, modulation of smooth muscle contraction, and secretion of neuropeptide. 73
The mammalian bombesin receptor family consists of four receptor subtypes: GRP receptor (BB2 or GRPR), neuromedin B receptors (BB1 or NMB-R), BB3, and BB4. 74
GRPR was found to be predominantly expressed in human prostate cancer (100%), gastrinoma (100%), breast cancer (70%) small and non small cell lung cancer (concomitant expression with BB3 receptor) gastrointestinal carcinoid tumors, and head/neck squamous cell cancers. 25,73 –75 Recently, Flores et al. confirm the presence of GRPR in human glioma specimens and normal human neurons. 76
BB1 receptors are overexpressed in small cell lung cancer (55%), non small cell lung cancer (67%), and intestinal carcinoids (46%). 73
Small cell lung carcinomas and bronchials carcinoids express the BB3 receptors. 25,74,76,77
Bombesin analogs for the GRPR have been successfully labeled with 68Ga. Some side effects have been reported with bombesin agonists, so it may be preferable to use antagonists for targeting GRPR positive tumors. 78,79
Zhang et al. have synthetized and radiolabeled a bombesin analog: [68Ga]-DOTA-PEG4-BN(7-14) (DOTA-PESIN). 80
Binding experiments, performed in vitro on human prostate tumors (PC-3 cells) show an IC50 of radiotracer of 6.6 nmol/L for human GRPR, revealing a good affinity for its biological target.
In vivo experiments, performed on PC-3 cells xenografts in athymic nude mice showed a high and specific tumor uptake and good retention. [68Ga]-DOTA-PESIN accumulates mainly in tumor, pancreas, and kidneys.
Others bombesin analogs have been successfully synthetized and radiolabeled with 68Ga such as [68Ga]-NODAGA-AR by Abiraj et al. 81 and [68Ga]-RM2 by Mansi et al. 82
Neurotensin and ductal pancreatic adenocarcinoma
Neurotensin is a brain gut 13 amino-acid peptide involved in neuronal functions, as neuromodulator in the central nervous system. In the periphery, neurotensin is released from entero-endocrine cells of the gastro intestinal tract in response to increased intraluminal fats, and modulates hormone activity involved in the digestive process, stimulation of pancreatic and biliary secretion, inhibition of gastric acid secretions. … 83 Neurotensin receptors (NTRs) family contains 3 subtypes: neurotensin receptor 1 (NTR1), neurotensin receptor 2 (NTR2), and neurotensin receptor 3 (NTR3). NTR1 and NTR2 are GPCR, respectively the high and low affinity neurotensin receptor. NTR3 is a nonspecific single trans-membranous sorting receptor.
It was found that ductal pancreatic adenocarcinomas express NTR1 (75–88%) while NTR1 is not detected in normal pancreas cells. NTR1 has been proposed as a marker of ductal pancreatic adenocarcinoma progression and as a discriminant between endocrine and exocrine pancreatic adenocarcinomas. 83
Moreover, invasive ductal breast cancers express NTR 1 (90%), while NTR1 is poorly detected in normal breast cells. 84,85 Numerous cancers express NTRs but with less frequency, such as Ewing's sarcoma (65%), meningioma (52%), and astrocytoma (43%). 86
Alshoukr et al. have synthetized and radiolabeled with 68Ga two DOTA-neurotensin analogs: [68Ga]-DOTA-NT-20.3 and [68Ga]-DOTA-LB119. In vitro assays on HT-29 cells (human colon adenocarcinoma cells) showed an IC50 of 14 and 7.5 nM (respectively) using [125I]-labeled neurotensin as competitor.
Biodistribution studies were performed on athymic mice, bearing a HT-29 cells xenografts (Fig. 8). 18 Tumor uptake of [68Ga]-DOTA-NT20.3 was superior to the [68Ga]-DOTA-LB119.
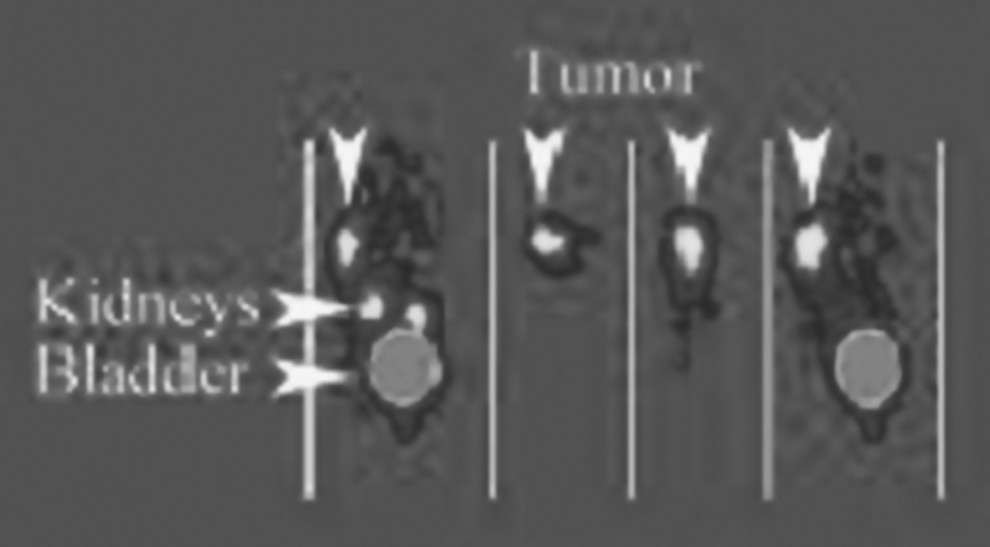
PET images of HT-29 cells xenografts. Image taken from Alshoukr et al. Reprinted with permission from American Chemical Society Copyright (2011).
Moreover, image contrast was better with [68Ga]-DOTA-NT20.3 than [68Ga]-DOTA-LB119 allowing the detection of small tumors.
Neuropeptide Y system and breast cancer
The neuropeptide Y system is composed of neuropeptide Y (NPY), peptide YY (PYY), and pancreatic polypeptide (PP). They act through their receptors named Y1, Y2, Y4, and Y5. There are all GCPR.
More specifically, NPY, a 36 amino-acid peptide is a sympathetic neurotransmitter and could also act as an angiogenic factor.
NPY receptors are expressed in three categories of cancer 87 : endocrine, 88,89 epithelial 90,91 , and embryonal tumors. 92 –94
In endocrine tumors, producing steroid hormones and cathecolamines, Y1 and Y2 subtypes are the most express among NPY receptors.
Epithelial tumors, and more particularly breast cancer, are the tumors expressing the highest NPY receptors density.
Embryonal tumors such as neuroblastoma express only Y2 and Y5 receptors whereas Ewing's Sarcoma family of tumors express Y1, Y4, and Y5 receptors. Density and incidence of NPY receptors are moderate in these tumors.
According to Korner and Reubi, breast cancer seems to be the best candidate for targeting NPY receptors based on their very high NPY receptors expression. 87
Until now, no NPY analogs has been radiolabeled with 68Ga or has successfully passed preclinical trials, so further optimization is needed even though neuropeptide Y system receptors are still promising targets.
“Glucagon-like”peptides and NET
“Glucagon-like” peptide 1 (GLP-1) and 2 are incretin hormones derived from proglucagon after enzymatic cleavage in pancreas. GLP-1 is also released in gut and brain. It is then metabolized into inactive metabolites by dipeptidyl peptidase 4 (DPP-4). GLP-1 plays an important role in metabolism and maintenance of glucose homeostasis. GLP-1 exerts its action via its receptor, GLP-1 receptor (GLP1R) belonging to GPCR superfamily. 95
It was shown that GLP1R is highly overexpressed in human insulinomas and gastrinomas. In insulinomas, GLP-1R density is considerably increased and is more frequently observed than somatostatin receptors. 96,97
Further, GLP1R is expressed in endocrine, neural, and embryonic tumors, whereas they are virtually absent in carcinomas and lymphomas. 77
Exendin-3 and GLP-1 have 53% of homology in their amino acid sequence. The main advantage of exendin-3 compared with GLP-1 is that exendin-3 is resistant to cleavage by the DPP-4.
Recently, [68Ga]-DOTA-Lys40-exendin-3 was synthetized, radiolabeled, and evaluated by Brom et al. for insulinomas detection. In vitro studies were performed on rat insulinoma cells INS-1 (GLP-1R positive). The results show an IC50 of 5.7 nM. Biodistribution studies in vivo were carried out on mice bearing xenografts of insulinomas INS1. High uptake of [68Ga]-DOTA-Lys40-exendin-3 was observed and the subcutaneous tumors were clearly visualized. 96
“Melanocyte-Stimulating” Hormones and melanoma
Melanocortin system is a network of skin neuropeptides involved in pigmentation and cortisol production regulation.
Natural Melanocyte Stimulating Hormones (MSH α, β, γ) and ACTH derive from pro-opiomelanocortin (POMC) by proteolysis under action of the prohormone convertase family. 98
There are five known melanocortin receptors called MSH receptors (MC-Rs) and belong to GPCR superfamily. There are also specific signaling pathways to each receptor.
Most cutaneous cells express MC-Rs and synthesize MCs, such as α-MSH. MC-1R is overexpressed in human and murine melanotic and amelanotic melanomas. 99,100
Several α-MSH analogs have been successfully synthetized and radiolabeled with 68Ga such as the [68Ga]-CHX-A″-ReCCMSH (Arg11) reported by Wei et al. This radiotracer specifically binds to MC-1R. In vitro binding experiments were performed on B16/F10 cells (murine melanoma) The IC50 of 3.8 nM suggests a very good affinity to its receptor. In vivo biodistribution experiments were carried out on C57BL/6 mice bearing a murine melanoma tumor xenograft B16/F10. The radiopharmaceutical is excreted in urine and has good tumor retention. PET imaging showed that tumor and urinary bladder are clearly visible. 101
“Gonadotrophin-Releasing” Hormone and cancers of the reproductive system
Gonadotrophin-Releasing Hormone (GnRH) is a hypothalamic hormone and plays an important role in reproduction. It is secreted in a pulsatile fashion by hypothalamus and binds to its receptor located mainly in the anterior pituitary gland to regulate production and release of luteinizing hormone (LH) and “follicle stimulating” hormone (FSH). LH and FSH control sex hormone production.
LH-RH receptors have been found to be expressed in most prostate cancer cells. 102 GnRH receptor exists in two isoforms called GnRHR-I and GnRHR-II. They are both GPCR. GnRHR has also been localized in tissues such as ovaries and breast and regulates menstrual cycle, pregnancy, and lactation. 103,104 Finally, GnRHR-I was detected in spleen and gastric parietal cells and plays a role in adhesion, chemotaxis of T cells, and inhibit acid gastric secretion. 105 GnRHR is highly overexpressed in tumors of reproductive organs such as endometrium, ovary, mammary gland, and prostate. 106
The GnRH-receptor system is suitable for in vivo peptide receptor targeting. Schottelius et al. have developed a GnRH-I analog radiolabeled with 68Ga, the (D-Lys6-[68Ga]-DOTA)-GnRH-I. 106
Competition studies were performed on cells membrane preparation expressing GnRHR and show an IC50 of 13.27 nM, revealing a good affinity for its biological target. However, this analog exhibits a loss of affinity close to 100 compared to the reference Triptoreline.
Conclusion
We can hypothesize that radiopharmaceuticals labeled with 68Ga have a promising future, especially 68Ga-labeled peptides due to the following reasons: (1) 68Ga half-life compatible with human PET-imaging; (2) daily production of 68Ga from a 68Ge/68Ga generator easy to manage in each PET imaging center; (3) peptide radiolabeling procedure.
With these undeniable advantages, several peptides involved in oncogenesis mechanisms have been successfully radiolabeled with 68Ga such as chemokine receptor CXCR4 ligand 107 or gastrin analog. 108 Nevertheless, other peptides involved in inflammation processes have been also radiolabeled with 68Ga to target leukocyte trafficking using Vascular Adhesion Protein 1. 20 Moreover, nonpeptide radiotracers labeled with 68Ga have been proposed for the exploration of P-glycoprotein expression, 109 hypoxia, 110 folate metabolism, 111 bone metastases, 112 and tumor metabolism. 113 Detection of gene mutations involved in oncogenesis is a way explored by Roivainen et al. thanks to radiolabeled oligonucleotides. 114
Macrocyclic chelators that form very stable complexes with metal cations are of paramount interest for radiopharmaceutical design. One advantage of chelate used for radioelement binding is that DOTA-based biomolecules can be labeled not only with 68Ga but also with nonradioactive gadolinium (Gd3+) giving access to the next generation of multimodal imaging that is PET-MRI. Boss et al. have used DOTA-TOC to detect meningioma with PET-MRI. 115 This technique has a huge potential for innovative imaging application by the combination of PET information (tracer uptake) and different MRI protocols (diffusion, functional, or spectroscopy imaging). 116
In addition, macrocycle chemistry allows also the attachment of other positron emitters (64Cu) or isotope for SPECT imaging (111In) or the setting of β-emitting isotopes for therapeutic peptides (coupled 86Y/90Y, 177Lu, and 67Cu). Labeling the same molecule with different radioisotopes (γ, β+, and β- emitters) will lead to development of novel innovative radiopharmaceuticals targeting specific molecular for both imaging and therapy applications. To conclude, molecular imaging of oncogenesis with radiolabeled peptides will hold an important place in the management of cancer, diagnosis and staging, using biochemical characteristics specific to each patient.
Footnotes
Disclosure Statement
No financial interests exist.