
Abstract
Second mitochondrial-derived activator of caspase (Smac) plays crucial roles in mitochondrial apoptosis pathways and promotes chemotherapy-induced apoptosis. In this study, Smac levels were examined in various lung cancer cell lines, and the effects of overexpressed Smac in the nonsmall-cell lung cancer cell line A549 were assayed by stable transfection of Smac. Subsequently, MTT assays, cell counting, and flow cytometry were applied to show that overexpression of Smac inhibits cell viability and cell growth and enhances apoptosis after cisplatin treatment. Western blotting was performed before and after cisplatin treatment to demonstrate that drug treatment could release Smac from mitochondria into the cytosol and promote apoptosis by activating caspase-3 and caspase-9. Promotion of apoptosis by cytosolic Smac could be blocked by pretreating cells with the caspase-9 inhibitor z-LEHD-FMK. Our findings indicate that overexpressed Smac significantly inhibited A549 cell growth and promoted apoptosis following cisplatin treatment due to the release of Smac from mitochondria into the cytosol, which increased the activities of caspase-3 and caspase-9.
Introduction
Non-small-cell lung cancer (NSCLC) accounts for nearly 80% of all lung cancers. Lung cancer is the leading cause of male cancer deaths and the second leading cause of female cancer deaths worldwide, resulting in a combined 18% (1.4 million) of cancer-related deaths in 2008. It is also one of the most commonly diagnosed cancers, and new cases of lung cancer account for 13% (1.6 million) of new cancer diagnoses each year. 1 In the United States, these percentages are even higher. Approximately 226,160 (13.8%) patients are diagnosed with lung cancer and 160,340 (27.8%) die annually. 2 The incidence of lung cancer is high, but the fatality rate is even higher due to the general ineffectiveness of conventional surgery and chemotherapy. 3,4
Platinum-based chemotherapy is internationally recognized as the first line therapy for patients with lung cancer. 5 However, cisplatin generates low success rates and, like many conventional, nontargeted chemotherapeutics, it produces severe side effects. 6 Therefore, new treatment regimens are required to increase the efficacy of cisplatin, enhancing antitumor effects while reducing dosage and side effects.
Over the past decade, much research has demonstrated that abnormal apoptotic pathways contribute to carcinogenesis and that the exploitation of these pathways for new cancer therapies would result in significant advances. 7 Second mitochondrial-derived activator of caspase (Smac) is a recently described component of the mitochondrial apoptosis pathway, whose expression is reduced in malignant tumors, 8 suggesting that Smac is a novel anticancer gene. Smac localizes to the mitochondria and is released into the cytosol in response to many apoptotic stimuli, such as chemotherapy and radiotherapy. 9,10 Upon release, Smac facilitates apoptosis induction by interacting with the inhibitor of apoptosis protein (IAP) and activating caspases. 9,11 Overexpression of Smac in cancer cells can sensitize chemotherapy-, radiotherapy-, and TRAIL-resistant cells to apoptosis. 12 –14
Smac mRNA expression has been reported to be significantly lower in lung cancer tissue than in normal lung tissue. NSCLC patients with higher Smac mRNA expression had significantly longer progression-free survival and overall survival with adjuvant chemotherapy. 15 –17 Therefore, we investigated whether overexpression of Smac could improve the efficacy of cisplatin. Here, we report that the ability of cisplatin to induce apoptosis was enhanced following overexpression of Smac in the A549 NSCLC line, and these cells showed increased activation of caspase-3 and caspase-9.
Materials and Methods
Construction of plasmids and transfection
Full-length Smac was generated by RT-PCR from total RNA extracted from human normal testis using the following primers: sense 5′-CGGGATCCCACAATGGCGGCTCTGA-3′ and antisense 5′-CCCAAGCTTGGCCCTCAATCCTCACGC-3′. The resulting 746-bp fragment was digested by BamHI and HindIII (Takara) and cloned into pcDNA3.1 (Invitrogen). The resulting plasmid or a control plasmid was transfected into NSCLC A549 cells by Lipofectamine 2000 (Invitrogen). Stably-transfected cells were selected in medium supplemented with 1 mg/mL G418 (Sigma) for 1 month. Western blotting was used to identify the cells with significant overexpression of Smac in A549 cells.
Cell culture and drug treatment
A549 NSCLC cells were maintained at 37°C and 5% CO2 in RPMI 1640 media (HyClone), supplemented with 10% FBS (HyClone), 100 units/mL penicillin, and 100 g/mL streptomycin. Cisplatin (Sigma) was diluted to the appropriate concentrations in cell culture medium before addition to cells. The caspase-9 inhibitor z-LEHD-FMK (Millipore) was diluted to a final concentration of 50 μM, and cells were pretreated 2 hours.
qPCR
After A549/Smac and A549/neo cells were treated with 10 μg/mL cisplatin for 24 hours, total RNA was extracted from cells using RNeasy Mini Kits (QIAGEN), and reverse transcription was performed with SuperScript III First-Strand Kits (Invitrogen). The Smac was amplified with SYBR green qPCR Master Mixture (Thermo) on a Lightcycler 480 II (Roche), with the following protocol: initial denaturation at 95°C for 15 minutes, then 45 cycles of 95°C for 10 seconds, 58°C for 30 seconds, and 72°C for 15 seconds. GAPDH gene was used as a control. The following PCR primer sequences were used for Smac: SMAC-F:ATCAGAGCCTCATTCCCTTAGT; SMAC-R: TGGTGTTTTGAAGTCATCTCAGC.
MTT assay
For cell viability assays, 5×103 A549 cells, with or without overexpression of Smac, were plated into each well of 96-well plates. The following day, different concentrations (0, 1.25, 2.5, 5, 10, 20, 40, and 80 μg/mL) of cisplatin were added to the wells for 24 hours. After 24 hours, 20 μL of MTT (Sigma) was added to each well for 4 hours. Then, the supernatant was discarded and 100 μL DMSO was added to each well, and the absorbance at 490 nm (A490) was measured with a Microplate Reader (Bio-Rad). Each cell viability assay was performed with five replicate wells. Cell viability (%) was calculated as follows: Cell viability=the average A490 in an experimental chemotherapy group/the average A490 in the blank control group ×100%.
Growth curves
After treating with 5 μg/mL cisplatin for 24 hours, A549/neo and A549/Smac cells were cultivated in 24-well plates (1×104 cells/well) for 1–6 days. Viable cells were distinguished from inviable cells by trypan blue, and the number of live cells was counted using a Countess Automated Cell Counter (Invitrogen). The growth curves use the average of triplicate samples for each time point.
Flow cytometry
Cells were plated into 24-well plates with a concentration of 1×105 per well. The following day, cisplatin was added to a final concentration of 0, 5, or 10 μg/mL. After 24 hours, cells were trypsinized and incubated with 5 μL propidium iodide and 5 μL Annexin V-FITC (Invitrogen) for 15 minutes. Samples were then analyzed for apoptosis by a FACScan flow cytometer.
Western blot analysis
Following cisplatin treatment, cells were scraped into lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 0.05% SDS, 1% Triton X-100) and incubated for 30 minutes on ice. Normalized protein amounts were separated on 12% SDS-PAGE gels and transferred to PVDF membranes (Millipore) by semi-dry blotting. After treatment with 5% BSA at room temperature for 1 hour, the membranes were incubated with anti-Smac rabbit antibody (1:1000; Abcam) overnight at 4°C, incubated with horseradish peroxidase labeled goat antirabbit IgG antibody (1:5000, Santa Cruz) for 1 hour at 25°C, and developed using ECL detection. For immunodetection, the following primary antibodies were used: anticaspase-9 and anticaspase-3 rabbit antibodies (1:1000; Cell Signaling Technology).
Results
Differential expression of Smac in lung cancer cells
To analyze the effect of Smac expression in cisplatin-treated lung cancer cells, we first sought to determine the Smac levels in various cell lines, including A549, H23, H157 (adenocarcinoma), U1810 (large cell cancer), and U1690 (small-cell cancer). In these five different cells lines, Smac expression was the lowest in A549 (Fig. 1), and this cell line was selected for further study.

Expression of second mitochondrial-derived activator of caspase (Smac) in lung cancer cell lines. β-actin was used as a loading control.
Overexpression of Smac inhibits cell viability and cell growth and enhances cisplatin-induced apoptosis
A549 cells were transfected with a control plasmid or pcDNA3.1-Smac and selected to generate stable cell lines (A549/neo or A549/Smac, respectively). Western blotting confirmed that the expression of Smac in A549/Smac was higher than either of the control cell lines (Fig. 2A).

Overexpression of Smac inhibits cell viability and cell growth and enhances cisplatin-induced apoptosis.
A549, A549/neo, and A549/Smac cells were treated with a range of concentrations of cisplatin (0, 1.25, 2.5, 5, 10, 20, 40, or 80 μg/mL) for 24 hours, and their viability was assessed by MTT assay, which showed dose-dependent viability. A549/Smac cells showed significant decreases in viability at low doses of cisplatin compared with the control A549/neo cells. There was no significant difference between the cell viability of A549 and A549/neo under any concentration of cisplatin (Fig. 2B). After treatment with 5 μg/mL cisplatin for 24 hours, the A549/Smac cells proliferated more slowly than A549/neo (Fig. 2C).
To examine the apoptotic cell death of A549/Smac cells treated with cisplatin, we treated A459/neo and A549/Smac cells with either 5 or 10 μg/mL of cisplatin for 24 hours. After 24 hours, the cells were stained with propidium iodide and FITC-Annexin V to assess apoptosis induction by flow cytometry (Fig. 2D). Untreated cell cultures showed similar low levels of apoptosis, but in cisplatin-treated cultures, the number of apoptotic A549/Smac cells was significantly higher than in A549/neo (Fig. 2E).
Cisplatin treatment releases Smac from mitochondria into the cytosol
Under normal conditions, Smac localizes to the mitochondria, but following apoptotic stimuli, it is released into the cytosol. Therefore, we examined the release of Smac following cisplatin treatment. A549/Smac cells were treated with 5 or 10 μg/mL cisplatin for 24 hours, and the expression of Smac in both mitochondria and the cytosol was analyzed by western blotting. The cytosolic Smac increased with a corresponding decrease in mitochondrial Smac during treatment of A549/Smac cells with cisplatin (Fig. 3).
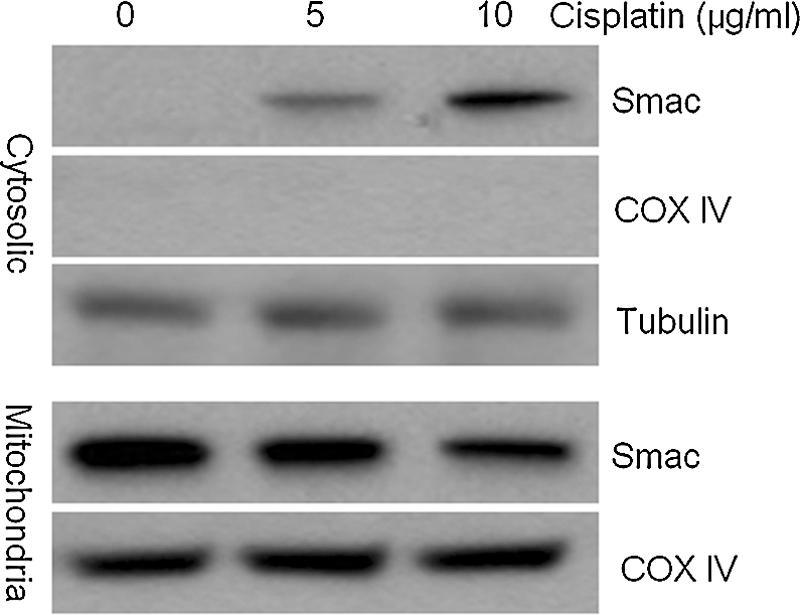
Smac was released from mitochondria into the cytosol following treatment with cisplatin. The expression of Smac in both mitochondria and the cytosol after cisplatin treatment was examined by western blot analysis. COX IV and Tubulin were used as loading controls.
Cytosolic Smac promotes apoptosis by activating caspase-3 and caspase-9
To investigate the increased induction of apoptosis in A549/Smac cells following cisplatin treatment, western blotting was performed for components of the apoptosis pathway. Following treatment with 10 μg/mL cisplatin, the expression of endogenous Smac was increased and the activities of caspase-3 and caspase-9 also increased. Notably, in A549/Smac cells, the activities of caspase-3 and caspase-9 were substantially increased following cisplatin treatment, which indicated that increased Smac expression promotes the activation of caspase-3 and caspase-9, resulting in increased apoptosis (Fig. 4A). Interestingly, in the absence of cisplatin treatment, the effect of Smac overexpression on caspase-3 and caspase-9 activation is not significant, possibly because Smac is properly located in the mitochondria in stably transfected cells and cannot spontaneously trigger in apoptosis. During treatment with 10 μg/mL cisplatin for 12 hours and 24 hours, Smac was released into the cytosol gradually, activating of caspase-3 and caspase-9 and promoting apoptosis (Fig. 4B).
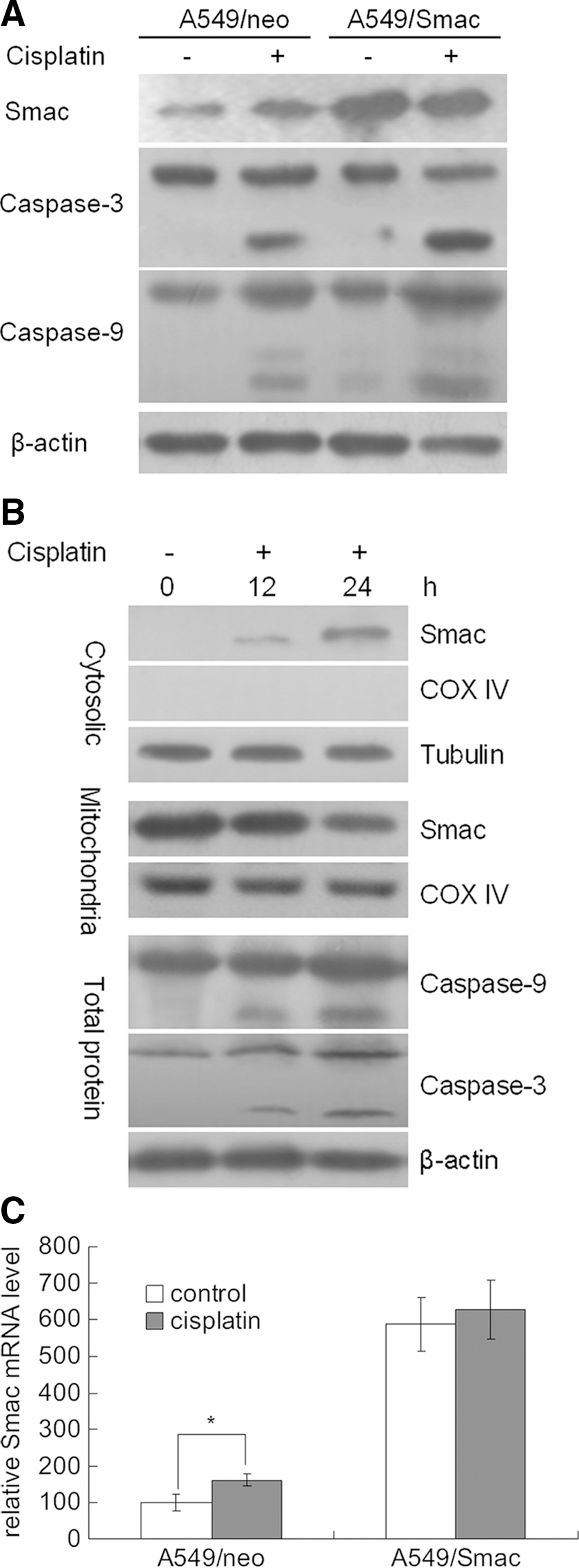
The increase of Smac expression in the cytosol promotes apoptosis by activating caspase-3 and caspase-9.
qPCR indicated that Smac mRNA levels were significantly increased in A549/neo cells during treatment with cisplatin. However, in A549/Smac cells, because of the expression of Smac from a CMV promoter, the mRNA and protein levels of Smac are saturated, independent of cisplatin treatment (Fig. 4C).
Caspase-9 inhibition blocks apoptosis induction by cisplatin
To investigate the requirement for caspase-3 and caspase-9 in cisplatin-induced apoptosis in Smac-expressing cells, A549/Smac cells were treated with 10 μg/mL cisplatin for 24 hours in the presence or absence of the caspase-9 inhibitor z-LEHD-FMK. During cisplatin treatment, the increase of Smac in the cytosol directly correlated with apoptosis induction and the activation of caspase-9 and caspase-3. The expression of cytosolic Smac did not change following treatment with z-LEHD-FMK, but the activation of caspase-9 and caspase-3 was reduced (Fig. 5A). Additionally, the number of apoptotic A549/Smac cells was significantly reduced following inhibition of caspase-9 (Fig. 5B). These data support the hypothesis that cytosolic Smac can activate caspase-9 and caspase-3 to induce apoptosis.
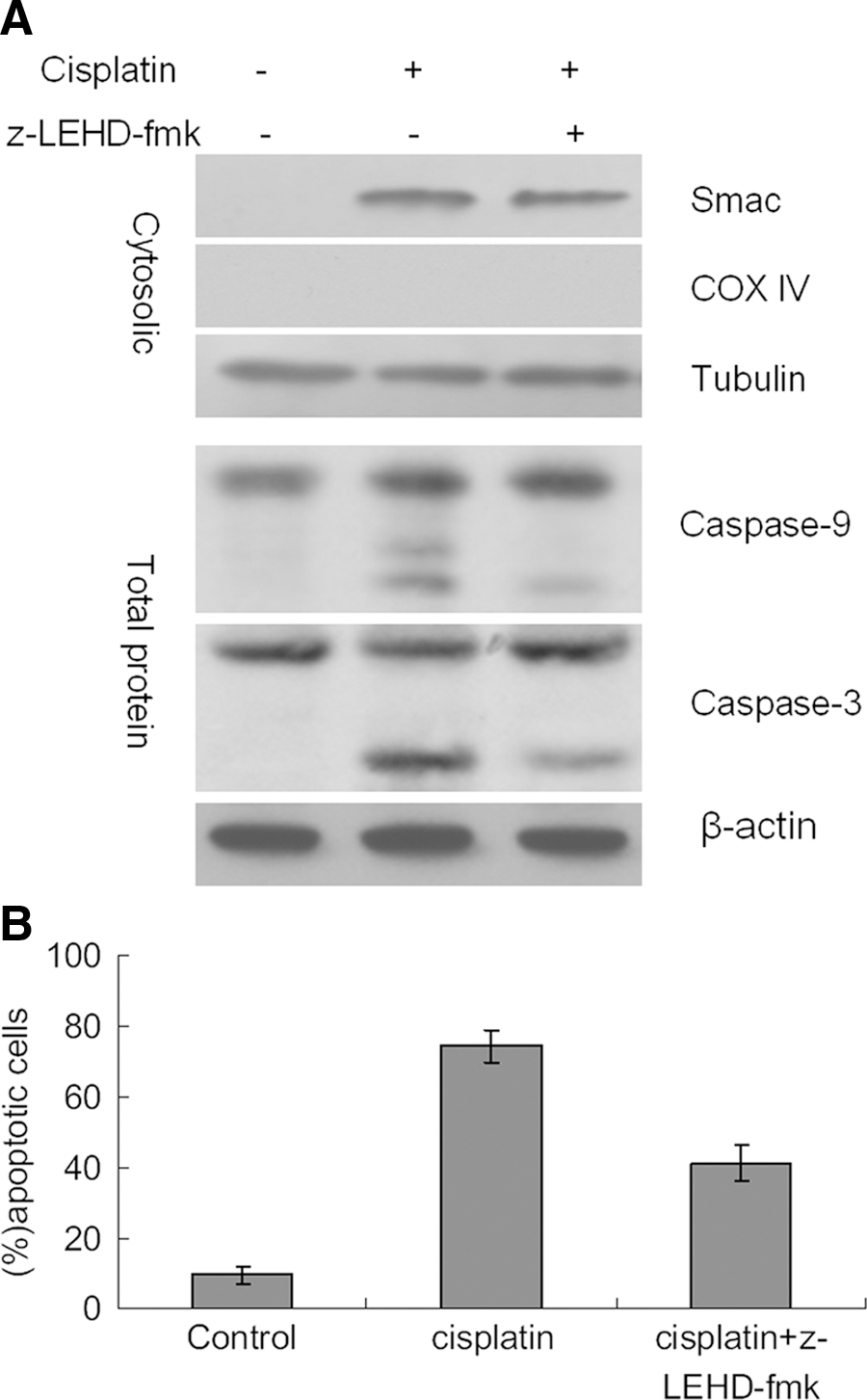
Inhibition of caspase-9 blocks induction of apoptosis.
Discussion
Cisplatin is one of the most widely used chemotheraptics and is a first-line drug for lung cancer treatment. However, high doses of cisplatin produce strong side effects on patients, such as severe renal, neurologic, and emetogenic effects, and low doses do not effectively curtail tumor growth. 18,19 Therefore, improvements of the standard cisplatin treatments should increase the antitumor effect at lower doses to reduce toxicity. 20 –22 Cisplatin is known to induce tumor cell apoptosis via the activation of caspases, and it is anticipated that modulation of apoptosis pathway components, such as the IAPs, could augment the effectiveness of cisplatin. 23,24
Smac was initially discovered by Du et al. 8 and Verhagen et al. 9 in 2000. Under apoptotic stimuli, it is released from mitochondria and translocates to the cytoplasm, where it binds IAPs, activates caspases, and promotes apoptosis in Hela cells. 8 We demonstrated that stable overexpression of Smac in A549 cells could not induce apoptosis because Smac does not promote apoptosis until it is released into the cytosol. During treatment of A549/Smac cells with cisplatin, cytosolic Smac levels increased, with a corresponding decrease in mitochondrial Smac. The percentage of apoptotic A549/Smac cells was higher than A549/neo cells after treatment with cisplatin. We conclude that, in response to cisplatin, Smac is released from mitochondria into the cytosol to promote apoptosis in A549 lung cancer cells.
Smac function is closely related to the IAP family members, XIAP, c-IAP1, c-IAP2, and Survivin. IAPs usually have three Baculoviral IAP Repeats (BIRs) and a really interesting new gene zinc finger domain. 25 BIR1, BIR2, and BIR3 domains are each capable of inhibiting caspases independently, and each also interacts with Smac. For example, in XIAP, the BIR2 domain and the linker between BIR1 and BIR2 are required for inhibition of caspase-3 and the XIAP BIR3 domain plays an important role in blocking caspase-9 activity. 26 We found that cisplatin treatment of Smac-overexpressing cells increased cytosolic Smac expression and resulted in increased activity of caspase-9 and caspase-3. The caspase-9 inhibitor, z-LEHD-FMK, could inhibit the activation of caspase-9 and caspase-3, but did not alter the expression of Smac, possibly because Smac binds BIR2 and BIR3 of XIAP and prevents XIAP from inhibiting the activity of caspase-9 and, subsequently, caspase-3 (Fig. 6). 27 –29
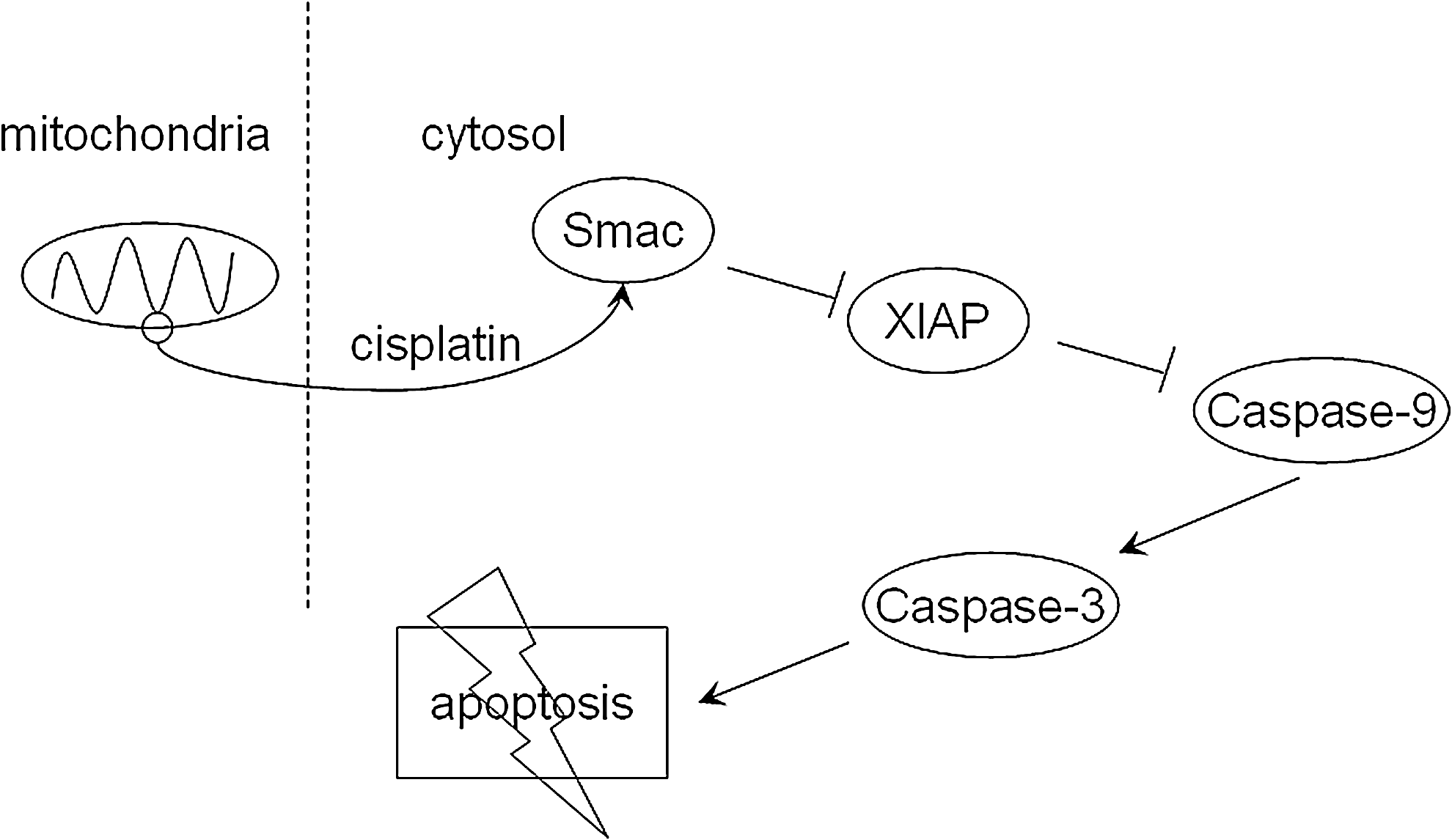
A model for Smac-mediated increases in cisplatin-induced apoptosis. Smac is released from mitochondria into the cytosol by treatment with cisplatin, where it could activate caspase-9 and caspase-3 by blocking XIAP, leading to apoptosis.
Smac has clear roles in the development of lung cancer and is an important prognostic indicator. Additionally, it may inhibit tumor growth and its pro-apoptotic effects make it an important target in the generation of targeted therapies. 30 Previous studies of Smac-mediated antitumor therapy have pursued two approaches: Smac upregulation in conjunction with chemotherapy 31 and/or radiotherapy 12,13 or administration of Smac peptide. 32 –35 However, the latter approach is compounded by new research that suggests exogenous Smac peptide is unstable in vivo. 36 Therefore, the expression level of Smac is increasingly becoming a primary focus of Smac-related therapies. Here, we have demonstrated that overexpression of Smac increases the sensitivity of lung cancer A549 cells to cisplatin treatment, and re-introduction of Smac to tumor cells represents one potential therapy. However, more research is required to precisely identify the effects of Smac in lung cancer models and to determine if there is synergy in combination with TRAIL, chemotherapy, or radiotherapy.
Footnotes
Acknowledgments
We thank J. R. Skaar for critically reading the article. This research work was supported by the Shannxi Province Science and Technology Fund (2010K01-133) and the Chinese National Natural Science Foundation (30740022).
Disclosure Statement
The authors declare that there are no financial conflicts of interest.