
Abstract
Different cyclooxygenase (COX)-2 inhibitors were known to cause different cell cycle changes. We investigated whether this different effect on cell cycle change was due to concentration-dependent effect. We investigated the effects of celecoxib, a COX-2 selective inhibitor, on cell cycle regulation in irradiated cancer cells that express high or low levels of COX-2. Four stably COX-2 knocked-down or overexpressed cell lines were treated with various concentrations of celecoxib with or without radiation. Celecoxib differentially modulated the cell cycle according to the concentrations applied. G1 arrest was induced at lower concentrations, whereas G2/M arrest was induced at higher concentrations in each cell line tested. Radiation-induced G2/M arrest was enhanced at lower concentrations but reduced at higher concentrations. The cutoff values to divide lower and higher concentrations were cell-type specific. Celecoxib treatment activated Cdc25C and inhibited p21 expression in both unirradiated and irradiated cells, regardless of COX-2 expression. Apoptosis was induced in irradiated cells 48 hours after treatment with celecoxib dependent of COX-2. These results imply that celecoxib deactivates the G2 checkpoint via both Cdc25C- and p21-dependent pathways in irradiated cells, which subsequently die by secondary apoptosis. Cell cycle modulating effects in irradiated cells resulting from treatment with celecoxib may have clinical importance with regard to the potential application of celecoxib in cancer patients undergoing radiotherapy.
Introduction
Cyclooxygenase (COX) is a key enzyme that catalyzes the conversion of arachidonic acid to prostaglandins (PG), and other prostanoids. Two COX isoforms have been identified. COX-1 is constitutively expressed in a variety of cell types and appears to be intimately involved in the homeostasis of several physiological functions, whereas COX-2 is an inducible enzyme that is regulated by various factors, including cytokines, growth factors, and tumor promoters. 1,2 Increased COX-2 expression has been observed in many tumor types in both humans and animals, and COX-2 selective inhibitors have been reported to prevent carcinogenesis and reduce the growth rate of tumor cells both in vitro and in vivo. 3,4 In addition, COX-2 selective inhibitors sensitize tumor cells to both chemotherapeutic agents and ionizing radiation. 5
COX-2 selective inhibitors are known to modulate the cell cycle in both normal and cancer cells. COX-2 selective inhibitors primarily induce G0/G1 arrest, 3,6 –11 but under certain conditions some COX-2 inhibitors have been shown to induce G2/M arrest. 12 –14 Although several researchers have studied the underlying mechanisms and conditions of cell cycle modulation by COX-2 selective inhibitors, it remains unclear whether such differential effects (induction of G0/G1 or G2/M arrest) are drug-specific or dependent on drug concentration. Further, little is known about the effects of COX-2 selective inhibitors on cell cycle regulation in irradiated cells. A well-known effect of ionizing radiation is profound cell cycle changes; therefore, potential modulation of these events by COX-2 inhibitors is of interest.
We previously reported that celecoxib, a COX-2 selective inhibitor, can radiosensitize cancer cells in a COX-2 expression-dependent manner, and that modulation of radiation-induced G2/M arrest may be an underlying mechanism of this effect. 15 In the current study, we investigated the concentration-dependent differential effects of celecoxib on cell cycle regulation in irradiated cells expressing high or low levels of COX-2 to further understand the mechanism of cell cycle modulation by COX-2 selective inhibitors and the interaction between COX-2 selective inhibitors and radiation in cancer cells.
Materials and Methods
Reagents
Celecoxib was provided by Pfizer Inc.
Cell culture
A549 human lung adenocarcinoma and HCT-116 human colon adenocarcinoma cells were acquired from the American Type Culture Collection and cultured in the recommended media supplemented with 10% fetal bovine serum, 50 units/mL penicillin, and 50 μg/mL streptomycin (all from Life Technologies). For all experiments, cells were cultured for no more than eight passages and only cultures that were less than 90% confluent were used.
Immunoblotting
Cells were lysed for 30 minutes at 4°C in radioimmunoprecipitation assay (RIPA) buffer [1×phosphate buffered saline (PBS), 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1 mM EDTA] containing 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 μg/mL pepstatin, and 100 μg/mL phenylmethylsulfonylfluoride. The protein concentration in the supernatant from centrifuged cell lysates was determined using a Bio-Rad Protein Assay Kit (Bio-Rad Laboratories) according to the manufacturer's instructions. Proteins were denatured and fractionated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The filters were incubated overnight at 4°C in blocking solution (PBS containing 5% nonfat, dried milk and 0.1% Tween 20), followed by incubation for 1 hour with primary antibodies. Primary antibodies were rabbit polyclonal anti-COX-2 (Cayman Chemical), and rabbit polyclonal anti-p27Kip1 (p27), anti-Cdc25C, anti-phosphorylated Ser216 Cdc25C (pCdc25C), anti-phosphorylated Tyr15 cyclin dependent kinase (Cdk)1 (pTyr15-Cdk1), anti-phosphorylated Thr161 Cdk1 (pThr161-Cdk1), anti-cleaved caspase-3, mouse monoclonal anti-p21Waf1/Cip1 (p21), anti-Cdk1, and anti-cyclin (Cell Signaling Technology). The filters were washed and incubated with horseradish peroxidase-conjugated immunoglobulins at a 1:5000 dilution as secondary antibodies for 1 hour. After additional washes, the filters were developed with an enhanced chemiluminescence system (GE Healthcare) and exposed to Hyperfilm ECL (GE Healthcare). The membranes were also probed with anti-actin antibody (Sigma) to normalize differences between the samples. Quantitation was conducted by video densitometry.
Development of stably COX-2 overexpressing or knocked down cells
We designed and developed a stably COX-2 knocked down cell line using vectors expressing COX-2-specific siRNA as previously described. 15 A stably COX-2 overexpressing cell line was also developed as previously described. 16
Detection of cell cycle changes and apoptosis by flow cytometry
In brief, 2.5–5×105 cells were plated into 25 cm2 flasks for the determination of each data point. After 24 hours, cells were exposed to the appropriate concentrations of celecoxib or vehicle (dimethyl sulfoxide [DMSO]) for 4 hours and then exposed to appropriate doses of γ-rays using the Gammacell 3000 Elan system (MDS Nordion, Inc.), at a dose rate of 10.7 Gy/min. After incubation for an additional 20 hours in medium containing either drug or vehicle, the cells were trypsinized (retaining all floating cells), fixed with 75% ethanol at 4°C overnight, and incubated at room temperature with 10 μg/mL propidium iodide (Sigma) and 5 μg/mL RNase A (Amresco) for 3 hours. The number of cells in each cell cycle stage, and the number of cells that had undergone apoptosis (sub-G1) was evaluated with the FACSCalibur system (Becton Dickinson). 17 Error bars were also calculated, as±standard error of mean (SEM), by pooling results of three independent experiments.
Statistical analysis
Data were expressed as mean±SEM and were analyzed with regard to statistical significance using analysis of variance, followed by Scheffe's test for multiple comparisons. A p-value <0.05 was considered significant.
Results
Cell cycle changes induced by various concentrations of celecoxib
A549 cells exhibit high constitutive COX-2 expression levels. A stably COX-2 knocked down A549 cell line (AS) and the corresponding control cell line (AN) were used in the present experiments. HCT-116 cells exhibit no constitutive COX-2 expression. A stably COX-2-overexpressed cell line (HCT-116-COX-2) and its empty vector control (HCT-116-mock) were used in the present experiments. HCT-116-mock/HCT-116-COX-2 cells were more sensitive to celecoxib or radiation than AN/AS cells. We therefore used lower doses of celecoxib or radiation in HCT-116-mock/ HCT-116-COX-2 cells than AN/AS cells to provide similar growth-inhibiting conditions with these treatments and to fairly compare cell cycle regulatory effects in both cell systems.
To characterize cell cycle changes after celecoxib treatment of cells expressing high or low levels of COX-2, we performed flow cytometric analyses of each cell cycle phase in each cell line after propidium iodide staining. Under basal condition, the proportion of cells in the G2/M phase was higher in AN- and HCT-116-COX-2 cells (AN cells; 6.9% and HCT-116-COX-2 cells; 21.5%) than in their control cell lines (AS cells; 5.3% and HCT-116-mock cells; 14.4%) (Fig. 1).

Cell cycle changes after treatment with celecoxib and/or radiation in AN
When AN- or AS cells were treated with various concentrations of celecoxib for 24 hours, 50 and 75 μM celecoxib induced G0/G1 phase prolongation, whereas 100 μM celecoxib induced S- and G2/M phase prolongation. Similarly, in HCT-116-COX-2 and HCT-116-mock cell lines, 40 or 60 μM celecoxib induced G0/G1 and synchronous G2/M phase prolongation, whereas 80 μM primarily induced G2/M phase prolongation. No significant differences were observed between the cells expressing high and low levels of COX-2 (Fig. 1). These results indicate that celecoxib differentially regulates cell cycle dependent on its concentration but regardless of COX-2 expression levels.
Cell cycle changes induced by various concentrations of celecoxib in irradiated cancer cells
Next, we investigated cell cycle changes by various concentrations of celecoxib treatment in irradiated cancer cells. At 24 hours following radiation treatment, the degree of radiation-induced G2/M phase prolongation was significantly higher in HCT-116-COX-2 cells than in HCT-116-mock cells (85.4% vs 61.7%) and slightly higher in AN cells than in AS cells (39.7% vs 37.0%) (Fig. 1).
Fifty micromolar celecoxib for AN/AS cells, and 40 or 60 μM for HCT-116-mock/ HCT-116-COX-2 cells resulted in enhanced radiation-induced G2/M phase prolongation in COX-2 low-expressing (AS and HCT-116-mock) cells, but no significant changes in G2/M phase were shown in COX-2 overexpressing cells. In contrast, treatment with higher concentrations of celecoxib in each cell line (75 or 100 μM for AN/AS cells and 80 μM for HCT-116-mock/ HCT-116-COX-2 cells) decreased the radiation-induced G2/M phase prolongation in all tested cells (Fig. 1). The degree of G2/M phase reduction was similar in AN and AS cells but was different in the HCT-116-mock and HCT-116-COX-2 cell lines; 8% reduction in G2/M arrest in HCT-116-mock and 17.9% in HCT-116-COX-2 compared with HCT cells treated with radiation alone (Fig. 1).
Regulation of molecules involved in the G2 checkpoint by treatment with celecoxib and radiation
Cdk1 and cyclin B1 are final effector molecules in the G2 checkpoint that form a complex to promote G2 to M transition. The activity of Cdk1-cyclin B1 complex is regulated by expression and phosphorylation status of Cdk1 and cyclin B1. 18 Therefore, to investigate the underlying molecular mechanism of G2/M phase regulation by celecoxib±radiation, the expression and phosphorylation status of Cdk1 and cyclin B1 were evaluated by Western blot analysis. AN/AS or HCT-116-mock/ HCT-116-COX-2 cells were pretreated with various concentrations of celecoxib for 4 hours, exposed to 9 or 6 Gy radiation, and further incubated for 20 hours for 100 or 80 μM celecoxib treatment, or up to 68 hours for 50 or 40 μM celecoxib treatment. The treatment with 100 or 80 μM celecoxib±radiation significantly reduced the protein levels of Cdk1 and Cyclin B1 compared with control or radiation alone in all four cell lines regardless of their COX-2 expression levels. Similarly, the phosphorylation of both inhibitory (Tyr-15) and activatory (Thr-161) residues of Cdk1 was inhibited by celecoxib treatment with or without radiation in all cell lines (Fig. 2). Similar finding was shown in AN/AS cells when treated with 50 μM celecoxib (at 24 hours) but the effect was more prominent in AN cells compared with AS cells (Fig. 2A). In HCT-116-mock/ HCT-116-COX-2 cells treated with 40 μM celecoxib, reduction of cyclin B1 expression and Cdk1 phosphorylation were shown only in HCT-116-COX2 cells at 72 hours after radiation (Fig. 2C). These results suggest that celecoxib causes downregulation of Cdk1 phosphorylation and cyclin B1 expression, and it is shown in a COX-2-dependent manner at 40–50 μM but independent of COX-2 at 80–100 μM.
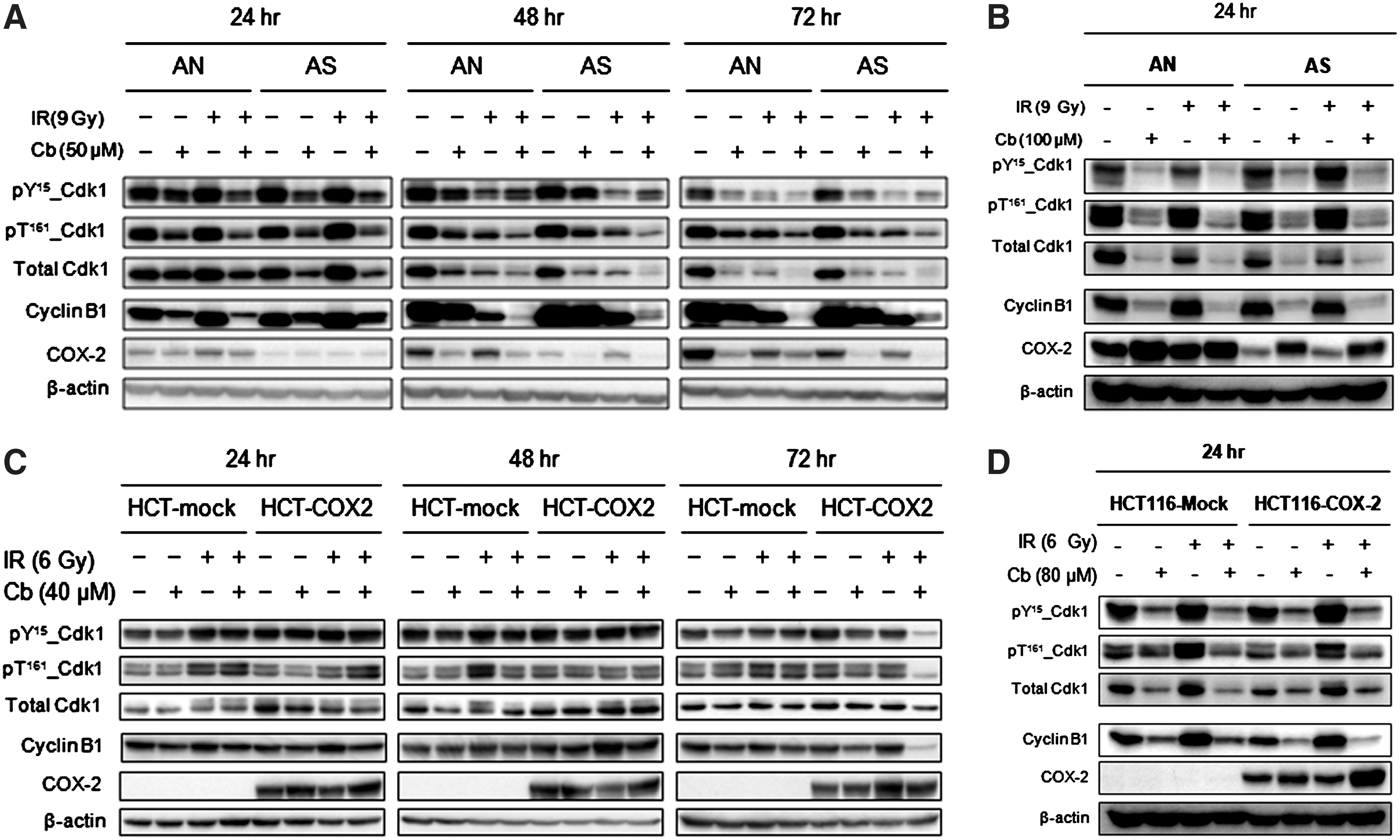
Western blot analysis for expression and phosphorylation of Cdk1 and cyclin B1 in irradiated or unirradiated cells expressing high or low levels of COX-2. AN/AS
Next, we evaluated regulatory molecules of the Cdk1-Cyclin B1 complex in the G2 checkpoint. The treatment with 100 or 80 μM celecoxib±radiation reduced the phosphorylation of Cdc25C at serine 216 residue and p21 protein expression compared with control or radiation alone in all four cell lines (Fig. 3). The treatment with 50 μM celecoxib±radiation showed similar results in AN/AS cells (Fig. 3A). In HCT-116-mock/ HCT-116-COX-2 cells treated with 40 μM celecoxib showed reduction of p21 expression in only irradiated cells, and the change in phosphorylation of Cdc25C at serine 216 residue was not significant (Fig. 3C).
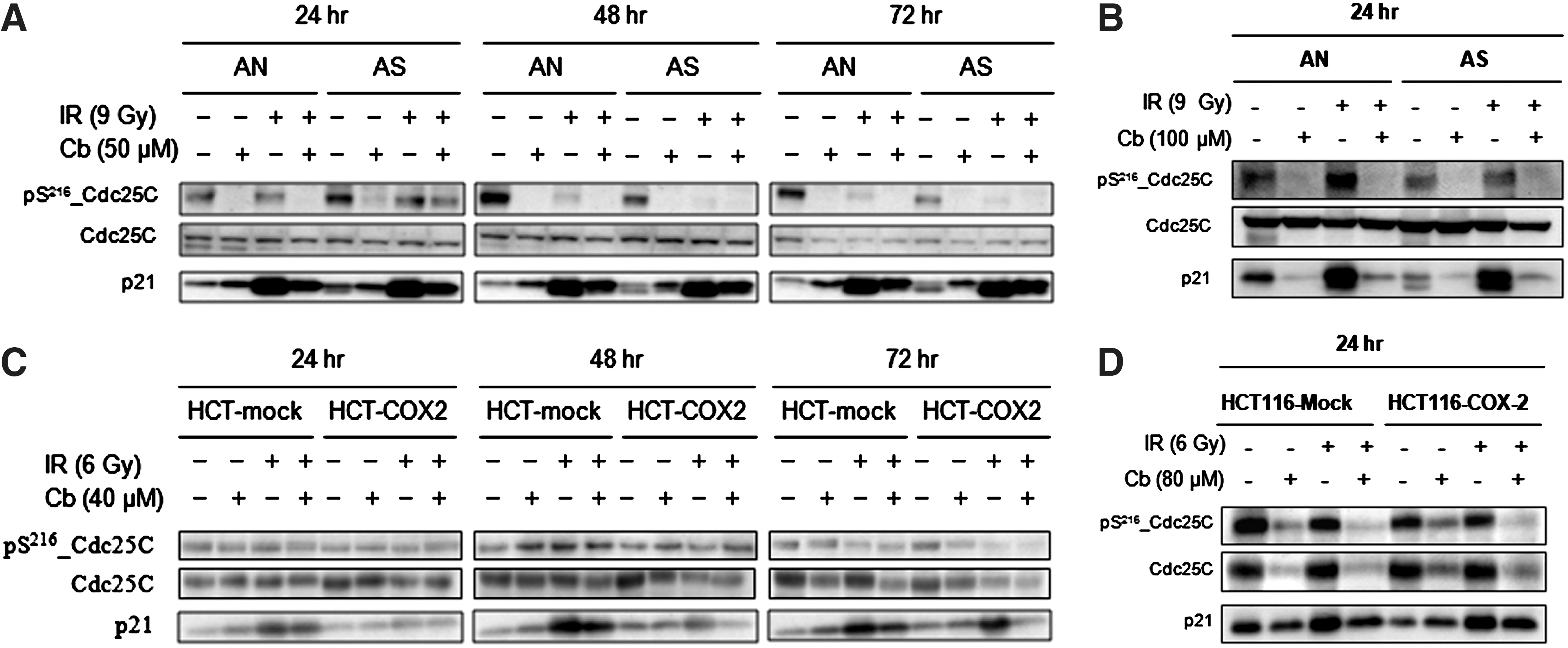
Western blot analysis for expression and phosphorylation of Cdc25C and p21 in irradiated or unirradiated cells expressing high or low levels of COX-2. AN/AS
Cooperative induction of apoptosis following combined treatment of celecoxib with radiation in COX-2 overexpressing cells
We investigated whether the observed cell cycle modulation by celecoxib eventually lead to increased apoptosis. In AN or AS cells, apoptosis was increased compared with control groups after treatment with 100 μM celecoxib for 24 hours, and there was no difference between the two cell lines (Fig. 4A, left panel). In contrast, a greater degree of apoptosis was induced in HCT-116-COX-2 cells after treatment with 80 μM celecoxib than in HCT-116-mock cells (Fig. 4B, left panel). Combined treatment with 100 or 80 μM celecoxib and radiation showed significantly enhanced apoptosis in all cell lines when compared with radiation alone. In addition, significantly higher levels of apoptosis were induced in AN and HCT-116-COX-2 cells than in AS and HCT-116-mock cells, respectively (Fig. 4A, B, right panels).
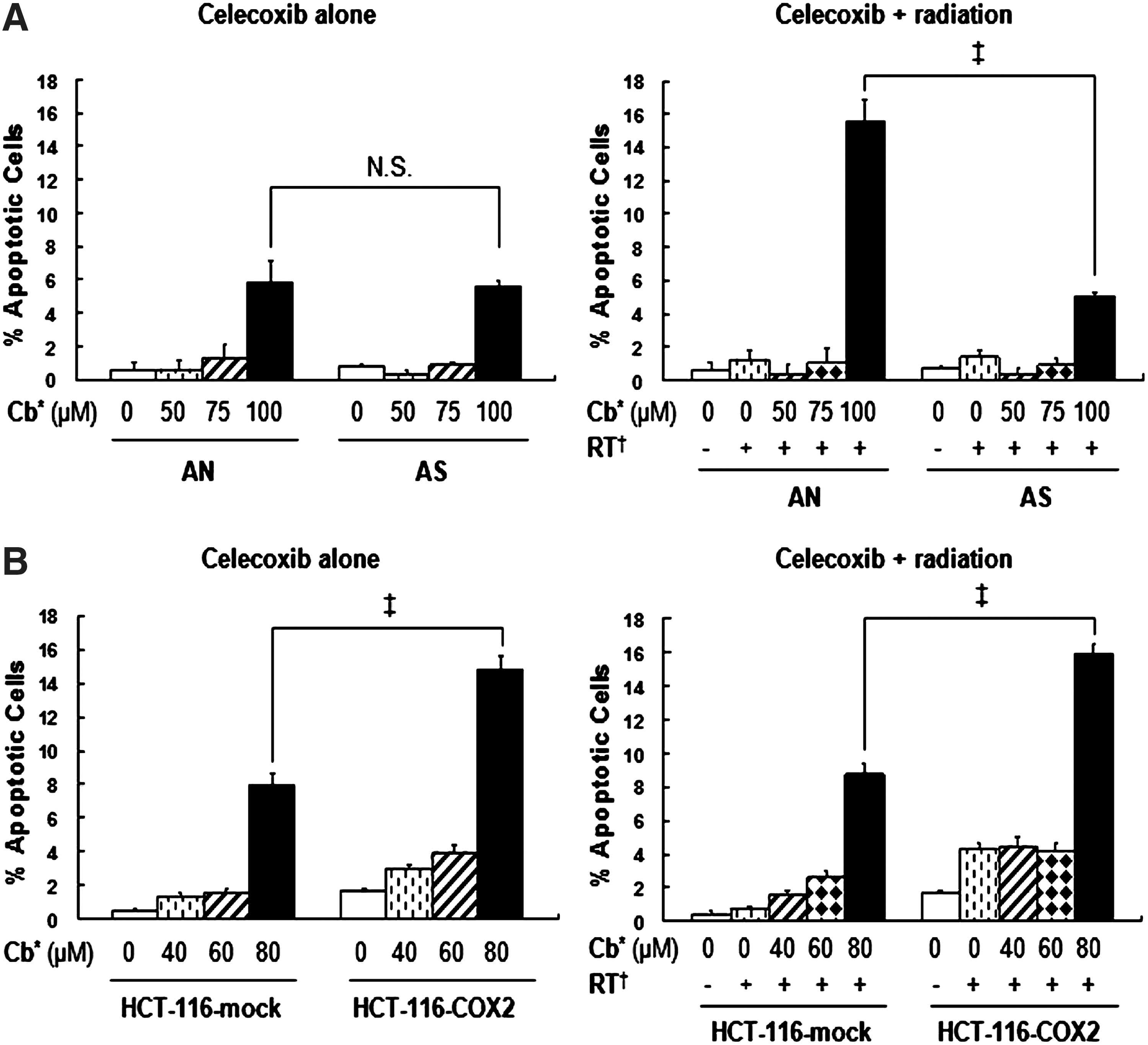
Apoptosis induction after celecoxib treatment either without (left panels) or with (right panels) radiation in AN and AS cells
To further verify this result, we measured the level of cleavage of caspase-3, a molecular marker of apoptosis. Cleavage of caspase-3 was not observed 24 hours after 100 or 80 μM celecoxib treatment with or without radiation in any of the cell lines (data not shown). However, cleavage of caspase-3 was evident after 48 hours of celecoxib treatment, alone or in combination with radiation, and the overall trend in caspase-3 activation was the same as that determined by flow cytometric analyses (Fig. 5).
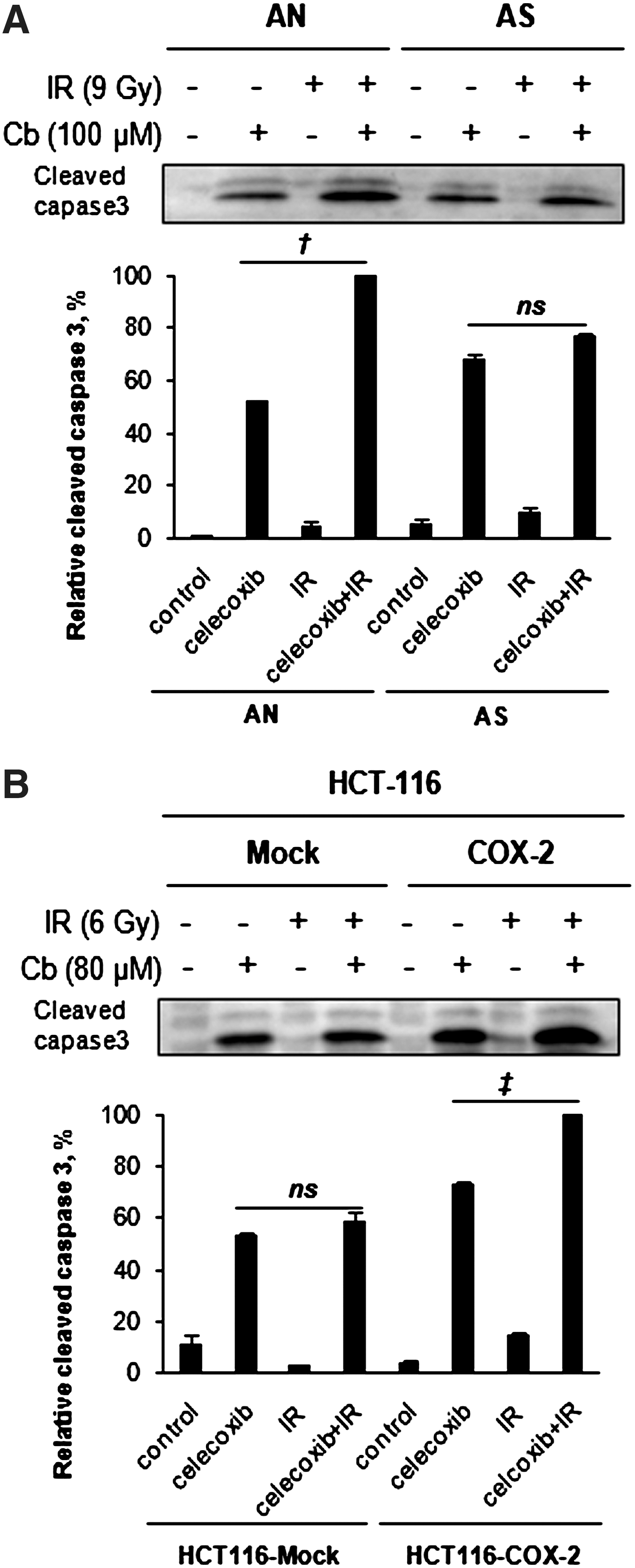
Detection of caspase-3 activation by Western blot analysis of cleaved caspase-3 in irradiated or unirradiated cells expressing high or low levels of COX-2. Cells were exposed to 100 μM or 80 μM celecoxib for 4 hours and then exposed to 9 or 6 Gy of radiation in AN/AS cells
Discussion
We showed in the current study that different concentrations of celecoxib result in different types of cell cycle changes in each cell line; lower concentrations induced G0/G1 arrest, whereas higher concentrations induced G2/M arrest in each cell line tested, although the cutoff values to divide lower and higher concentrations were cell-type specific. These effects were independent of COX-2 expression level. Current results indicate that cell cycle regulation by celecoxib occurs differentially according to the concentrations applied. This may be a reason for the different cell cycle regulations by different COX-2 inhibitors in the literature, and further studies using several concentrations for each COX-2 inhibitor may confirm this hypothesis.
We also found that different concentrations of celecoxib result in different cell cycle changes when the treatments are combined with radiation. Lower concentrations of celecoxib induced minor changes in radiation-induced G2/M arrest in COX-2 over-expressing cell lines, but enhanced radiation-induced G2/M arrest in COX-2 low-expressing cell lines (Fig. 1). In contrast, celecoxib treatment at higher concentrations reduced radiation-induced G2/M arrest in each cell line regardless of the COX-2 expression levels. Of note, eighty micromolar celecoxib reduced radiation-induced G2/M arrest to a greater degree in HCT-116-COX-2 cells than in their mock control cells. These results indicate that celecoxib modulates cell cycle differentially in both drug concentration and COX-2-dependent manners in irradiated cells. These COX-2-dependent differential effects were not observed between AN and AS cells, and the explanation for this may be incomplete knock down of the COX-2 protein in AS cells.
Radiation commonly induces G2 arrest in cancer cells, and this radiation-induced G2 arrest is caused by activation of the G2 checkpoint in an effort to protect cells against radiation damage. 19,20 Since high concentrations of celecoxib reduced radiation-induced G2/M arrest, we evaluated several molecules involved in the G2 checkpoint after treatment with celecoxib at these concentrations combined with radiation to determine whether celecoxib modulates the G2 checkpoint pathways to cause a reduction in G2/M arrest.
Cdk1 and cyclin B1 are two final effector molecules in the G2 checkpoint that form a complex to initiate progression from G2 to the mitotic phase. The activity of the Cdk1-cyclin B1 complex is regulated mainly by a Cdc25C-involved (p53-independent) pathway and also by a p53/p21-dependent pathway. 18 –21 We found that celecoxib treatment, alone or in combination with radiation, activated Cdc25C via reduction of its inhibitory phosphorylation at Ser 216, and also inhibited the expression of p21 in all cell lines. Both activation of Cdc25C and inhibition of p21 cause activation of the Cdk1-cyclin B1 complex, thereby promoting G2/M transition. 18 –21 These results imply that celecoxib inhibits the radiation-activated G2 checkpoint through both Cdc25C- and p53/p21-dependent pathways, and this effect may have caused the reduction in radiation-induced G2 arrest. Deactivation of the G2 checkpoint after celecoxib treatment may force irradiated cells to progress into mitosis without appropriate G2 arrest, thereby causing an early reduction of Cdk1 and cyclin B1 expression due to their early exit from mitosis 22 compared with cells subjected to radiation only. At lower concentrations, these effects were shown in a COX-2-dependent manner.
The irradiated cells with G2 checkpoint deactivation by celecoxib appeared to progress into mitosis and die by secondary apoptosis when considering delayed caspase-3 activation (at least after 24 hours). These effects were shown in a COX-2 expression-dependent manner in all cell lines tested (Fig. 5). Notably, although cell cycle regulation after celecoxib treatment in irradiated AN/AS cells was COX-2-independent (Fig. 1), apoptosis induction was dependent on COX-2 expression in these cell lines (Fig. 5). Therefore, there may be additional COX-2-dependent steps between cell cycle modulation by celecoxib and induction of secondary apoptosis.
These results support that celecoxib can act as a “G2 checkpoint inhibitor” to enhance the effect of radiation (reviewed in ref. 23,24 and see the discussion in ref. 15 ). However, the concentrations used in the current experiments are too high to be achieved in vivo 25,26 and alternative approaches may be needed to apply the current results to the treatment of cancer patients undergoing radiotherapy. Such approaches may include the administration of physiologically achievable lower concentrations of celecoxib over a longer time period, since the effect of a drug is usually proportional to both applied concentration and time.
It is puzzling that celecoxib treatment inhibited the G2 checkpoint in both unirradiated and irradiated cells but enhanced G2/M arrest in unirradiated cells, whereas reduced G2/M arrest in irradiated cells. In contrast to irradiated cancer cells that are mainly arrested in G2 phase by the G2 checkpoint activation, unirradiated cancer cells are mainly in G0/G1 or S phase. Celecoxib may activate the Cdk1-cyclin B1 complex in unirradiated cells via the same pathway as in irradiated cells. However, if the Cdk1-cyclin B1 complex is prematurely activated during G0/G1 or S phase, the complex may degrade or be suppressed rather than leading cells to progression into the G2 and mitotic phases. This suppression of Cdk1 and cyclin B1 may subsequently lead to G2 arrest due to a lack of active effector molecules for the G2-M transition. Another possibility is that celecoxib may also modulate other Cdk-cyclin complexes and therefore its effect is cell-cycle phase-specific. Extensive studies on cell cycle regulation by celecoxib, including the use of cell cycle-synchronized cells, are needed to clarify these complicated observations.
In summary, we showed that celecoxib differentially modulates cell cycle regulation according to the concentrations applied. Treatment with higher concentrations of celecoxib seems to deactivate the G2 checkpoint via both Cdc25C- and p53/p21-dependent pathways in irradiated cells, subsequently causing COX-2-dependent apoptosis. Despite the limitation of using nonphysiological high concentrations of celecoxib for short lengths of time, our observations may enhance our understanding of the basic mechanisms of cell cycle regulation by celecoxib at various concentrations and under various conditions.
Footnotes
Acknowledgments
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (KRF-2009-0074782) and by a grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (2010-1037-000).
Disclosure Statement
There are no existing financial conflicts.