
Abstract
Purpose:
Pretargeted radioimmunotherapy (PRIT) against intraperitoneal (i.p.) ovarian microtumors using avidin-conjugated monoclonal antibody MX35 (avidin-MX35) and 211At-labeled, biotinylated, succinylated poly-
Methods:
Mice were inoculated i.p. with 1×107 NIH:OVCAR-3 cells. After 3 weeks, they received PRIT (1.0 or 1.5 MBq), RIT (0.9 MBq), or no treatment. Concurrently, 10 additional animals were sacrificed and examined to determine disease progression at the start of therapy. Treated animals were analyzed with regard to presence of tumors and ascites (tumor-free fraction; TFF), 8 weeks after therapy.
Results:
Tumor status at baseline was advanced: 70% of sacrificed animals exhibited ascites. The TFFs were 0.35 (PRIT 1.0 MBq), 0.45 (PRIT 1.5 MBq), and 0.45 (RIT). The 1.5-MBq PRIT group exhibited lower incidence of ascites and fewer tumors >1 mm than RIT-treated animals.
Conclusions:
PRIT was as effective as RIT with regard to TFF; however, the size distribution of tumors and presence of ascites indicated that 1.5-MBq PRIT was more efficient. Despite advanced disease in many animals at the time of treatment, PRIT demonstrated good potential to treat disseminated ovarian cancer.
Introduction
Ovarian cancer is characterized by late and unspecific symptoms. At the time of diagnosis, the majority of affected women have advanced disease and poor prognosis, with tumor spread beyond the ovaries. Because the disease disseminates primarily to the abdominal cavity, an effective intracavitary treatment of micrometastases is desired, preferably in a stage with minimal tumor burden after cytoreductive surgery and chemotherapy. Radioimmunotherapy (RIT) with the α-particle emitter astatine-211 (211At) has proven successful in preclinical studies, 1 –3 and in 2009, Andersson et al. completed a phase I study of 211At-labeled MX35 F(ab′)2 fragments for intraperitoneal (i.p.) RIT in patients with ovarian cancer in Gothenburg, Sweden. 4 However, results from preclinical studies indicate that the therapy could be improved further, as the penetration of labeled antibodies into tumors with diameters >0.1–0.2 mm appears restricted or slow. 5 For such tumors, a smaller molecule might offer better delivery of radioactivity than labeled antibodies, and pretargeted radioimmunotherapy (PRIT) is an interesting option, especially for short-lived α-particle emitters. 6,7 Although several pretargeting regimens have been assessed in preclinical and clinical studies, to our knowledge, the possible benefits of PRIT in the peritoneal setting have not yet been evaluated. 8 –12
We have previously performed in vivo distributions of 125I-labeled avidin-conjugated monoclonal antibody (MAb) MX35 (125I-avidin-MX35) and 211At-labeled, biotinylated, and succinylated poly-
The current study aimed to compare the therapeutic efficacy and toxicity of the described pretargeting system with that of conventional RIT using 211At-labeled MX35 against ovarian microtumors in nude mice.
Materials and Methods
General
The α-particle-emitting radionuclide 211At was produced at the Cyclotron and PET Unit, Rigshospitalet (Copenhagen, Denmark), via the 209Bi(α,2n)211At reaction. After transportation to the Department of Nuclear Medicine at the Sahlgrenska University Hospital (Gothenburg, Sweden), the 211At in the irradiated target was isolated as previously described. 14 After dry distillation, the radionuclide was dissolved in chloroform, which was evaporated under gentle nitrogen flow before labeling. An ionization chamber (Capintec; CRC-15 dose calibrator) was used to measure radioactive samples with activities of >100 kBq. Samples with activities of <10 kBq were measured using an NaI(Tl) γ-counter (Wallac, Wizard 1480).
The targeting agent MX35 is a murine IgG1 MAb directed toward a membrane transporter molecule (NaPi2b) that is expressed homogeneously in ∼90% of human epithelial ovarian cancers. 15 The hybridoma was obtained from the Ludwig Institute for Cancer Research and cultured at the Department of Cell and Molecular Biology at the University of Gothenburg. The human ovarian cancer cell line NIH:OVCAR-3 was obtained from the American Type Culture Collection (ATCC) and cultured at the Department of Oncology at the Sahlgrenska University Hospital. After i.p. inoculation, the tumor growth mimics the human disease, with peritoneal carcinomatosis and development of ascites. 16
For the effector molecule, CASLO Laboratory ApS synthesized poly-
Chemistry
To synthesize the pretargeting molecule, avidin-MX35, sulfo-SMCC-activated avidin was conjugated to thiolated MX35, and the resulting monomer fraction (MW ∼220 kDa) was isolated according to a previously described protocol. 17
The effector molecule (211At-B-PLsuc) was synthesized by first biotinylating poly-
Briefly, NHS-dPEG®
4-Biotinidase-resistant biotin was added at fivefold molar excess to poly-
211At-labeling of MX35 was performed according to a two-step method developed previously. 18 First, m-MeATE in DMSO was added in 7.5-fold molar excess to MX35 in 0.2 M carbonate buffer (pH 8.5). After 30 minutes of incubation, the conjugate was isolated in 0.2 M acetate buffer (pH 5.5) using a Sephadex NAP-5-column. Of the resulting m-MeATE-MX35, 125 μg was added to a vial containing 47 MBq of 211At oxidized by 10 μL of NIS in methanol/1% acetic acid (67 μM) during vigorous agitation. After 1 minute, 5 μL of NIS (1 mg/mL) was added; the reaction mix was incubated for 1 minute before 5 μL of sodium ascorbate (50 mg/mL) was added, and the 211At-MX35 was isolated using a Sephadex NAP-5-column.
The respective radiochemical purities (RCPs) of the 211At-B-PLsuc and 211At-MX35 products were evaluated by instant thin-layer chromatography and methanol precipitation. The binding of the effector molecule to NIH:OVCAR-3-cells pretargeted with avidin-MX35 was assessed in vitro using a previously described cell assay, as was the cell binding of direct-labeled MX35. 17
Animals and study groups
All animal experiments were performed according to protocols approved by the ethics committee of the University of Gothenburg. Nude mice (BALB/c nu/nu, Charles River) were used for the experiments.
After 1 week of acclimatization, the animals (5–6 weeks of age) were inoculated i.p. with 1×107 NIH:OVCAR-3 cells in 0.2 mL of cell medium. After 3 weeks, the animals received PRIT (two groups), RIT, or no treatment (PBS). At the same time, a group of 10 additional mice was sacrificed and examined with regard to tumor status. For PRIT, all animals received 25 μg (0.1 nmol) of avidin-MX35 in 0.75 mL PBS 24 hours before injection of 211At-B-PLsuc. Regarding the effector molecule, either 1.0 MBq/0.4 μg (designated the PRIT 1 group) or 1.5 MBq/0.6 μg (designated the PRIT 2 group) was administered in 0.75 mL PBS. For RIT, the mice were given 0.9 MBq/4 μg of 211At-MX35 in 0.75 mL PBS, and the nonirradiated control group received 0.75 mL PBS. All injections were administered i.p. to n=20 mice per group, and the mice were fed a biotin-deficient purified diet (Purina Mills, LLC) from 7 days before to 1 day after the injection of radiolabeled molecules or PBS.
To reduce nonspecific uptake of free 211At, sodium perchlorate hydrate (NaClO4·xH2O; Sigma-Aldrich) was administered twice before the injection of radioactivity. The animals were i.p. injected with 2.5 mg of sodium perchlorate in 0.1 mL of PBS (∼1.2 μmol/g body weight) at −24 hours and −1 hour. After therapy, the status of the animals was followed daily, and individuals with signs of high tumor burden or poor general condition were euthanized.
Eight weeks post-therapy, all remaining mice were sacrificed and dissected. The presence of ascites and macroscopic tumors was assessed by ocular inspection without treatment information. In addition, microscopic tumor growth was evaluated by light microscopy examination (magnification up to 120×) of biopsy samples from peritoneum, i.p. fat, diaphragm, and small intestine. For microscopic examination, formalin-fixed and paraffin-embedded samples were sectioned to a thickness of 4 μm at two levels 100-μm apart, and stained with hematoxylin and eosin.
In addition to the therapies, different activities of either 211At-B-PLsuc (1.0 MBq/0.4 μg; 1.5 MBq/0.6 μg) or 211At-MX35 (0.9 MBq/4 μg) were administered i.p. in 0.75 mL of PBS to tumor-free mice to assess myelotoxicity by analyzing the resulting depression of white blood cells (WBC), platelets (PLT), and hemoglobin (HGB) after 5, 14, and 21 days for each individual (n=5 animals per activity group). Biotin-deficient diet and blocking agent were provided as described above. Two control groups (n=5 in each) that received 0.75 mL of PBS were also followed according to the same scheme; one of these groups was administered a biotin-deficient diet and the other received a standard diet.
Dosimetry
The amount of tumor cells in the peritoneal cavity for ascites-bearing animals was approximated from measurements on ascitic fluid. The number of decays per cell was then estimated by dividing the number of immunoreactive radiolabeled vectors by the total number of tumor cells, assuming instant and stable binding of the labeled substances to cells. Using a cell size for NIH:OVCAR-3 previously estimated by Palm et al., 19 mean absorbed doses from specific irradiation in the conducted therapies were then analytically estimated for isolated cells using a cell-level dosimetry code. 20
Results
Chemistry
The degree of biotinylation of the polymer was assessed using 4′-hydroxyazobenzene-2-carboxylic acid (HABA) analysis and found to be approximately three biotin moieties per poly-
Therapeutic outcome
Of the 10 animals sacrificed at the time of therapy (3 weeks after cell inoculation), seven had developed ascites. In the three mice without ascites, tumor growth was confirmed by light microscopy. The results from the different therapy regimens are shown in Table 1, including the number of mice with tumor growth, ascites, and the resulting tumor-free fractions (TFFs) for the study groups. The microscopy findings were divided based on the tumor size: one group had tumors ranging from single cells to cell clusters of ∼1 mm in diameter, and one group had tumors >1 mm in diameter. Tumors larger than 1 mm were observed in 40% of the animals treated with PRIT 1 or RIT, and in 15% of animals treated with PRIT 2. Among PRIT 2-treated animals with tumor growth observed by microscopy, 73% (8/11) had tumors ranging in size from single cells to 1 mm in diameter. Among RIT-treated animals, the corresponding proportion was 27% (3/11). According to the one-sided Fisher exact test, the number of tumors >1 mm was significantly higher in the RIT group compared with the PRIT 2 group (p<0.05).
Tumor-free fraction (no macroscopic or microscopic tumors or ascites).
One mouse without signs of tumor growth or ascites was sacrificed prematurely because of deterioration.
TFF, tumor-free fraction; PRIT, pretargeted radioimmunotherapy; RIT, radioimmunotherapy; PBS, phosphate-buffered saline.
Forty percent (8/20) of RIT-treated animals, 15% (3/20) of PRIT 1-treated animals, and 0% (0/20) of PRIT 2-treated animals had ascites at the time of dissection. The difference in ascites incidence between RIT- and PRIT 2-treated animals was significant according to the two-sided Fisher exact test (p<0.05). All animals in the control group developed tumors and ascites except for one mouse, which was excluded because it was sacrificed prematurely at 5 weeks post-therapy due to general deterioration.
Dosimetry
Measurements on ascites-bearing animals indicated that the tumor cell concentration in the ascitic fluid may have been as high as 3×108 cells/mL at the start of therapy. Together with an estimation of the ascites volume (0.3 mL), a rough approximation of the total number of tumor cells in the peritoneal cavity at the time of irradiation was derived, ending up at around 1×108 cells per mouse for the current model. The amount of radiolabeled molecules per cell was estimated to 300–500, resulting in a mean absorbed dose from specific irradiation of <10 Gy for animals treated with RIT or PRIT 1, and ∼15 Gy for PRIT 2-treated animals.
Myelotoxicity
The results of the myelotoxicity study are depicted in Figure 1. No difference was observed between the two control groups that were provided with different diets. Therefore, their hematology data were pooled for the analysis, resulting in a single control group (n=10). WBC and PLT were significantly reduced (p<0.05) at day 5 postinjection (p.i.) for the irradiated groups compared with the control group. The lowest mean WBC count at day 5 p.i. was recorded for the 1.5-MBq 211At-B-PLsuc group, whose leukocytes were suppressed by 93%. At day 21 p.i., all animals in that study group had recovered their initial baseline WBC count. Corresponding WBC reductions were 78% for the 1.0-MBq 211At-B-PLsuc group and 89% for the 0.9-MBq 211At-MX35 group. For PLT, the suppression at 5 days p.i. was 31% for 1.0 MBq 211At-B-PLsuc, 42% for 1.5 MBq 211At-B-PLsuc, and 36% for 0.9 MBq 211At-MX35. No effect on HGB was observed for any group.
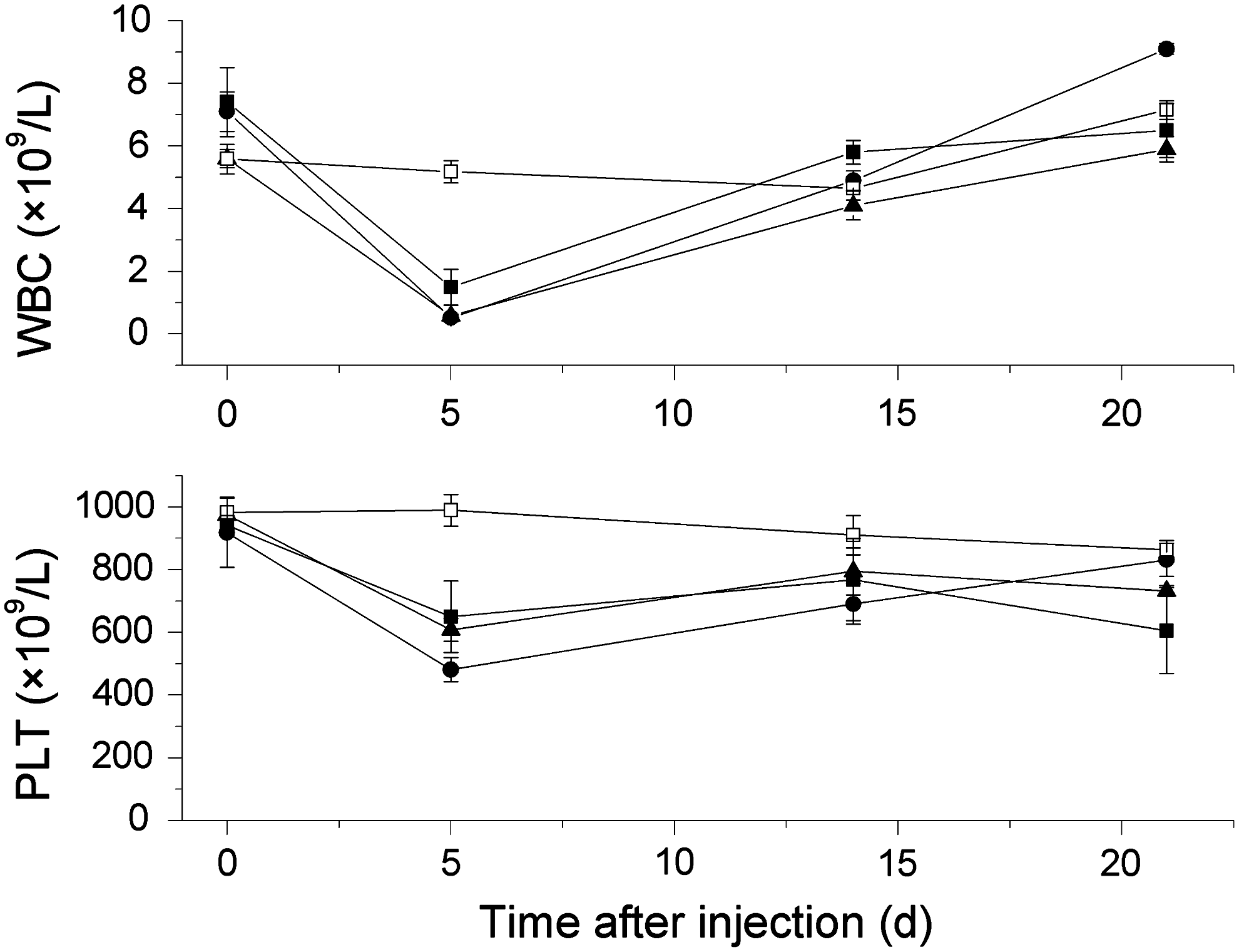
White blood cell (WBC) and platelet (PLT) counts as a function of time after intraperitoneal injections of 211At-B-PLsuc (1.0 MBq, ▪; 1.5 MBq, •; n=5), 211At-MX35 (0.9 MBq, ▴; n=5), or PBS (□; n=10). Values are means±SEM. A significant reduction in WBC and PLT counts (p<0.05) was recorded at day 5 postinjection for the irradiated groups compared with the nonirradiated control group.
Discussion
Several preclinical studies have demonstrated the efficacy of 211At-RIT for the treatment of ovarian carcinoma. 1,3,21 The encouraging TFFs (0.33–0.93) led to the realization of a phase I study with i.p. administration of 211At-labeled MX35 F(ab′)2-fragments against micrometastases in the abdominal cavity. Andersson et al. concluded from the pharmacokinetic data that potentially therapeutic absorbed doses could be delivered without any observed toxicity and with very low absorbed doses to bone marrow. 4 To continue that promising work, a phase II–III study is presently being planned. Still, in preclinical studies, tumors with a radius >0.1 mm have appeared less curable with α-RIT, possibly because of insufficient penetration of the labeled antibodies throughout the tumor volume. 1,5 Ideally, only microscopic residual disease remains in the peritoneal cavity after cytoreductive surgery, but in clinical reality, tumors may range from single cells to clusters of millimeter size. A pretargeting strategy might improve the labeled antibody distribution, thereby achieving a deeper and more uniform absorbed dose distribution also in the larger tumors. The pretargeting molecule can be given sufficient time to diffuse deep enough into the tumors, after which a small effector molecule can quickly find its way to the pretargeted tumor cells and deliver the therapeutic absorbed dose. This hypothesis remains to be thoroughly examined experimentally, which we aim to do in the near future using the novel alpha-camera technique that enables visualization and quantification of the activity distribution within tumors. 22 With PRIT, Pagel et al. observed indications of a favorable activity distribution as early as 45 minutes after the injection of 213Bi-labeled effector molecule using a mouse myeloid leukemia xenograft model. 23 Unpublished data from systemic biodistributions in our laboratory comparing 211At-RIT and 211At-PRIT in subcutaneous NIH:OVCAR-3 tumors also show greatly improved penetration of labeled molecules after pretargeting, already at 30 minutes p.i., supporting the findings by Pagel et al. Another benefit of pretargeting is the possibility of improving the therapeutic window by reducing the normal tissue irradiation. 24,25
In this study, PRIT using avidin-MX35 and 211At-B-PLsuc was compared with RIT using 211At-MX35. Even though a majority of the previous 211At-MX35 studies have been performed using F(ab′)2 fragments, full immunoglobulin G (IgG) was used to achieve comparable targeting with the IgG-based pretargeting molecule. Elgqvist et al. have previously shown that there is no evident activity–response relationship for i.p. 211At-RIT using administered activities of 0.1–1.2 MBq, possibly because of limited diffusion of the labeled antibodies in the tumors in combination with the short range of the α-particles. 1,2 In our RIT regimen, we thus aimed for 1.0 MBq, which is a tolerable activity for acute myelotoxicity in mice after i.p. administration (the maximum level being ∼1.2 MBq). 26 For PRIT, two different activity levels were chosen: 1.0 and 1.5 MBq. We have previously concluded that the bone marrow is most likely to be dose limiting for this pretargeting system due to an unexpected accumulation of radioactivity after the administration of 211At-B-PLsuc. Even so, data regarding bone marrow uptake and WBC depression after irradiation indicate that an injected activity of 1.0 MBq should deliver a tolerable absorbed dose to the mice. 13 Although unpublished results from further studies have suggested that the mice would be able to tolerate higher activities, there is no knowledge regarding the therapeutic effect of an increase in activity for i.p. PRIT (i.e., whether higher TFF could be reached, or whether the outcome would be relatively independent of the activity level, as in the case of i.p. RIT). The 1.5-MBq therapy group was added to the study to examine that possibility.
As expected, the irradiated animals experienced acute toxicity with regard to WBC and PLT counts at day 5 p.i., after which they recovered to normal levels within 21 days. According to previous studies, 1.0 MBq of i.p.-injected 211At provides an absorbed dose to the bone marrow of 0.9 Gy for 211At-B-PLsuc 13 and ∼2.5 Gy for 211At-MAb. 27 The administered activities would therefore correspond to 0.8 and 1.3 Gy for the groups treated with PRIT 1 and PRIT 2, and 2.1 Gy for the RIT-treated group. It should be noted that no conclusions can be drawn from this study regarding long-term toxicity effects, which deserve specific attention in future studies.
Although the three therapy regimens used here yielded similar TFFs, the outcomes differed between the groups at a more detailed level. The number of animals with macroscopic tumors observed at dissection was similar for PRIT 1-treated (1.0 MBq; 3/20), PRIT 2-treated (1.5 MBq; 1/20), and RIT-treated (0.9 MBq; 3/20) groups, as was the overall result regarding tumors observed by microscopy. However, when the microscopic findings were differentiated with regard to tumor size, a majority of the tumors found in the RIT-treated group were >1 mm, whereas few larger tumors were found in the PRIT 2-treated group. Another difference in the therapeutic outcome was the presence of ascites at the time of dissection: ascites was confirmed in 40%, 15%, and 0% of the animals treated with RIT, PRIT 1, and PRIT 2, respectively. Importantly, 7 of the 10 animals sacrificed at the time of irradiation, displayed ascites, indicating that a large proportion of study animals were at an advanced disease stage at the start of therapy. Elgqvist et al. have shown that for this in vivo model, the tumor status at the start of therapy strongly influences the therapeutic outcome for 211At-RIT, and the current TFFs correspond to those recorded for treatment initiated 5–7 weeks after cell inoculation. 5 The observed occurrence of ascites after therapy is interesting; if the initial ascites incidence was around 70% (as predicted by the sacrificed animals), the status was clearly improved 8 weeks post-treatment for the two PRIT groups, especially PRIT 2, while the change for the RIT group was more moderate. Although the TFFs were equal for RIT and PRIT 2, the ascites incidence and the distribution of tumor sizes in the two groups indicate that the PRIT 2-treated mice received more efficient therapy. One possible explanation for the observed effect is that better penetration, and therefore a more uniform absorbed dose distribution, was achieved for the larger tumors using PRIT. Other factors that are important regarding the therapeutic outcome are the amount of 211At-labeled molecules and the specificity of the irradiation. For small tumors, nonspecific irradiation with 211At has been shown to suffice; however, for larger clusters, the nonspecific absorbed dose contribution decreases with increasing tumor size, and the importance of a specific treatment increases drastically. 5
The total number of tumor cells is an important parameter that should be considered regarding how many targeting molecules to inject. According to previous studies performed in Gothenburg, the number of available antigens per cell is ∼1×106 for NIH:OVCAR-3 cells. 5 With the estimated 1×108 cells per mouse, roughly 1×1014 antigens were available in the peritoneal cavity at the beginning of therapy, while the number of antibodies and pretargeting molecules injected was in the order of 1×1013. This antigen excess over targeting molecules may have reduced the efficacy of the treatments by limiting the number of binding sites for the radiolabeled vectors, thereby reducing the mean absorbed doses delivered to tumor cells. It is important to stress that the calculated mean absorbed doses (<10 Gy for RIT and PRIT 1, and ∼15 Gy for PRIT 2) are relevant only for isolated cells or cells located on the edges of metastases; at this point, we were unable to estimate the mean absorbed dose received by tumor cells within metastases. For larger tumors and tumor clusters, some cells may have received no irradiation (i.e., 0 Gy). Further calculations indicated that the absorbed dose contribution from nonspecific irradiation deriving from radiolabeled vectors attached to free-floating tumor cells in the ascitic fluid was theoretically significant. However, for larger tumors (as were observed in some of the sacrificed animals at the beginning of therapy), this dose would have no effect on cells within the tumor core because of the limited range of the α-particles.
The amount of antibodies and pretargeting molecules administered thus turned out to be too small to cover all of the available antigens for the advanced disease in this study. Consequently, injecting a greater amount of the targeting substances should result in better TFFs for animals with ascites in the context of either RIT or PRIT. In addition, the dose estimations show that higher amounts of activity should be administered to increase the absorbed doses to tumor cells; however, the maximum tolerable activity is naturally restricted by uptake in normal tissues. Another option for increasing the absorbed dose is to optimize the ratio between pretargeting molecules and effector molecules, as shown by Sharkey et al. 28 In our therapies, the molar amount of avidin-MX35 was approximately twice that of 211At-B-PLsuc, a ratio that could be improved by increasing the specific activity of the effector molecule. However, increasing the specific activity of the effector molecule would require a method that enabled isolation of the labeled polymer fraction from the unlabeled fraction. This is virtually impossible using the present route of synthesis, because of the close chemical resemblance of the labeled and unlabeled fraction, that is, the 211At-B-PLsuc and the I-B-PLsuc. In addition, increasing the activity used for labeling beyond the levels presented using the current method is not an option, as the absorbed dose would be detrimental for the product. For prospective pretargeting studies, we therefore aim to modify the labeling procedure to enable optimization regarding the specific activity of the effector molecule, 211At-B-PLsuc.
Conclusions
Even though the comparison of therapeutic efficacy using 211At-PRIT and 211At-RIT in ascites-free animals remains to be evaluated, this study demonstrated proof of concept for the i.p. PRIT approach against ovarian carcinoma. PRIT using avidin-MX35 and 211At-B-PLsuc was equal to conventional RIT using 211At-MX35, in terms of TFF. However, accounting for the size distribution of tumors and the presence of ascites, the 1.5-MBq PRIT was shown to be somewhat more efficient than RIT. The TFFs were moderate overall, as the advanced degree of disease at the time of irradiation adversely influenced the therapeutic outcome. The high tumor burden at the start of therapy for many of the animals made the number of targeting molecules per cell inadequate, resulting in low absorbed dose to the tumors from specific irradiation. The larger tumor sizes also limited the impact of nonspecific irradiation, and reduced the accessibility to tumor cells to the radiolabeled vectors. More 211At was administered during the PRIT strategies than during RIT, resulting in higher specific absorbed dose, which, together with the possibility of better penetration, may explain the observed differences in therapeutic outcome.
In conclusion, we have shown that i.p. 211At-PRIT is a possible strategy for therapy of disseminated ovarian cancer. The studied pretargeting system was found to have good potential, even for animals with a large tumor burden that are generally difficult to treat with 211At-RIT. In future studies, the amount of pretargeting molecule and specific activity of the effector molecule should be optimized to increase the absorbed dose delivered to tumors.
Footnotes
Acknowledgments
The authors are highly grateful to Helena Kahu and Elisabet Warnhammar at the Department of Oncology, University of Gothenburg, Sweden, for skillful support regarding animal experiments and tumor cell culturing. We would also like to acknowledge Dr. Håkan Andersson and Ulla Delle for valuable contributions.
This work was supported by grants from the Swedish Cancer Society, the King Gustaf V Jubilee Clinic Research Foundation in Gothenburg, Sweden, the Swedish Research Council, and The Foundation Assar Gabrielsson for Clinical Research, specializing in cancer diseases.
Disclosure Statement
The authors declare that they have no conflicts of interest.