
Abstract
Hepatocellular carcinoma is the most common type of liver cancer. Radiotherapy combined with chemotherapy is the treatment of choice for hepatocellular carcinoma, but radioresistance of the cancer remains a significant therapeutic hindrance. Here, we provided several lines of evidence that small ubiquitin-like modifier (SUMO)-specific protease 6 (SENP6) could be an attractive molecular target for the treatment of hepatocellular carcinoma. By using immunohistochemical and real-time PCR, we showed that SENP6 was overexpressed in more than half of the hepatocellular carcinoma tissues. The growth retardation and radiosensitization were caused by silencing of SENP6 in the hepatocellular carcinoma cell lines using lentiviral shRNA. Moreover, SENP6 was required for radiation-induced NF-κB activation and the half-life of IκBα, a well-known inhibitor of NF-κB, and was extended by SENP6 silencing. Thus, our data demonstrated that SENP6 is an attractive drug target for anticancer therapy and radiosensitization.
Introduction
Small ubiquitin-like modifier (SUMO) proteins are a family of small proteins that are covalently attached to target proteins and detached from other proteins in cells to modify their function, and this process is called SUMOylation. 1 SUMOylation is a post-translational modification involved in various cellular processes. 2,3 Mammals express four SUMO isoforms: SUMO1, SUMO2, SUMO3, and SUMO4. SUMO2 has a 95% sequence homology with SUMO3. 4 SUMO conjugation was found to occur through a cascade of reactions performed by an activating enzyme (E1), a conjugating enzyme (E2), and a SUMO ligase (E3). 5 Before conjugation took place, SUMO must first be proteolytically processed from a proform by the action of a SUMO-specific protease (SENP) at the carboxy-terminus to expose 2 glycine residues and generate an active SUMO. 6,7 The SENPs exhibited differences in this endopeptidase activity. SENP1 and SENP2 expressed all three isoforms efficiently; SENP3 and SENP5 expressed the activity of only SUMO2 and SUMO3; and SENP6 and SENP7 did not demonstrate the hydrolase activity. 8
SUMOylation is also a dynamic, reversible covalent modification, and SENPs are responsible for deconjugating SUMO from target proteins. 9 SENP6 has been reported to be a nuclear protein localized to the nucleoplasm and preferentially acted on the substrates bearing the SUMO-2/3 chains. 10 Silencing of SENP6 caused the redistribution of SUMO2 and SUMO3 into the promyelocytic leukemia bodies. 11,12 Cells lacking SENP6 showed defects in the spindle assembly and metaphase chromosome congression. 13 In addition to these functions, a specific role of SENP6 has also been found in the regulation of the RPA complex and in initiating a Rad51-dependent homologous recombination. 14 However, whether SENP6 played a role in the development of cancer, especially hepatocellular carcinoma, was completely unexplored.
The NF-κB family of transcription factors includes the critical regulators of proinflammatory and antiapoptotic gene transcription programs. There are two signaling pathways leading to the activation of NF-κB known as the canonical pathway and the noncanonical pathway. 15 –17 The common regulatory step in both of these cascades is activation of an IkB kinase (IKK) complex consisting of catalytic kinase subunits (IKKa and/or IKKb) and the regulatory nonenzymatic scaffold protein NEMO (NF-kappa B-essential modulator). 18 Activation of NF-κB is due to IKK-mediated phosphorylation-induced proteasomal degradation of the IkB inhibitor, enabling the active NF-κB transcription factor subunits to translocate to the nucleus and induce target gene expression. 19,20 The NF-κB pathway is often hyperactivated in human cancers, including hepatocellular carcinoma. 21 –24
Here, we showed that SENP6 was overexpressed in human hepatocellular carcinoma tissues. Silencing of SENP6 slowed down the growth of a series of hepatocellular carcinoma cell lines. Moreover, SENP6 silencing enhanced radiosensitivity of hepatocellular carcinoma cells by blocking radiation-induced NF-κB activation.
Materials and Methods
Tissue samples
Primary hepatocellular carcinoma tissues and adjacent normal tissues were collected from a routine therapeutic surgery at our department. All samples were obtained with informed consent and were approved by the Changzheng hospital clinic institutional review board.
Immunohistochemistry
The 4-mm-thick fraction mounted on charged slides were immunohistochemically analyzed and sectioned from each clinical sample. Then, each slide was deparaffinized, and was treated with xylene and graded alcohol. After the antigen retrieval and being blocked with 5% bovine serum albumin, tissue slides were immunohistochemically stained by an antibody against SENP6, and then were visualized by a standard avidin–biotinylated peroxidase complex method.
Cell culture and drugs
Hepatocellular carcinoma cell lines, including HepG2, Hep3B, and SK-Hep1 (from the Cell Bank of Shanghai Institutes of Biological Sciences, Shanghai, China), were cultured in an RPMI-1640 medium (Sigma) supplemented with 10% fetal calf serum. All these cells were cultured in a 5% CO2/95% air at 37°C. DMSO and drugs in this work were purchased from Sigma.
Radiation exposure
Cells were seeded into 96-well plates and were treated with a range of radiation doses using a 250-kV orthovoltage unit the following day (Philips).
Antibodies
Antibodies were obtained from the following sources: anti-SENP6 (Santa Cruz Biotech), anti- IκBα (Cell Signaling), and antiactin (Sigma).
Transfection
Lipofectamine 2000 (Invitrogen) was used for the transient transfections according to the manufacturer's instructions.
RNA interference
Control and SENP6-shRNA lentivirus plasmids were purchased from Santa Cruz.
RNA isolation and real-time PCR analysis
The total RNA was isolated from tissues or cells using TRIzol (Invitrogen) according to the manufacturer's instructions. The transcripts of the interested genes were quantified by real-time PCR using a SYBR Green Premix Ex Taq (Takara) on LightCycler 480 (Roche). The primers used were available upon request.
Clonogenic assay
Cells were seeded into six-well plates after silencing of lentivirus-based shRNA. The next day, the cells were exposed to different doses of radiation and incubation for 7 to 9 days. The colonies formed were fixed, and the surviving fraction was the proportion of seeded cells irradiated to form colonies and untreated cells as described.
Luciferase reporter assay
HepG2 cells were infected with a lentivirus targeting SENP6, along with the control virus. Forty-eight hours after infection, the cells were seeded into a 96-well plate and were transfected with a luciferase reporter driven by an NF-κB consensus sequence (pNifty plasmid; Invivogen), along with a Renilla construct. Twenty-four hours after transfection, the cells were exposed to 10-Gy X-ray. The cells were harvested 24 hours later for the luciferase activity assay (Promega). The results were presented as the fold activation after normalization with Renilla.
Western blotting
Protein extracts were equally loaded on a 10%–12% SDS-PAGE, electrophoresed, and transferred to a nitrocellulose membrane (Amersham Bioscience). After blocking with 5% nonfat milk in PBS, the membranes were incubated with the indicated primary antibodies and followed by horseradish peroxidase (HRP)-linked secondary antibodies. The signals were detected by a chemiluminescence phototope-HRP kit (Pierce Biotechnology) according to the manufacturer's instructions.
Statistical analysis
Values were shown as mean±SD. The statistical differences were determined by a Student's t-test. The statistical significance is displayed as *p<0.05, **p<0.01, or ***p<0.001.
Results
SENP6 is overexpressed in human hepatocellular carcinoma tissues
Given the role of SENP6 in a DNA damage-response control, it should be necessary to check whether SENP6 played a role in tumorigenesis. First, we employed immunohistochemical staining with an anti-SENP6 antibody to detect SENP6 protein expression in a series of tissue sections of 40 hepatocellular carcinoma tissues and adjacent normal tissues. The results showed that SENP6 was highly expressed in 25 out of these 40 hepatocellular carcinoma tissues, but was hard to detect in their paired adjacent normal tissues (Fig. 1A). Then, the mRNA levels of SENP6 were detected in tumors and their adjacent normal tissues from 16 hepatocellular carcinoma samples by real-time quantitative PCR, and relative folds of SENP6 mRNA of tumor tissues against their adjacent tissues were calculated. As shown in Figure 1B, the SENP6 mRNA level in half of the cancer tissues was twofold higher than that of the paired adjacent normal tissues. Together, these data indicated that SENP6 was overexpressed in human hepatocellular carcinoma tissues.
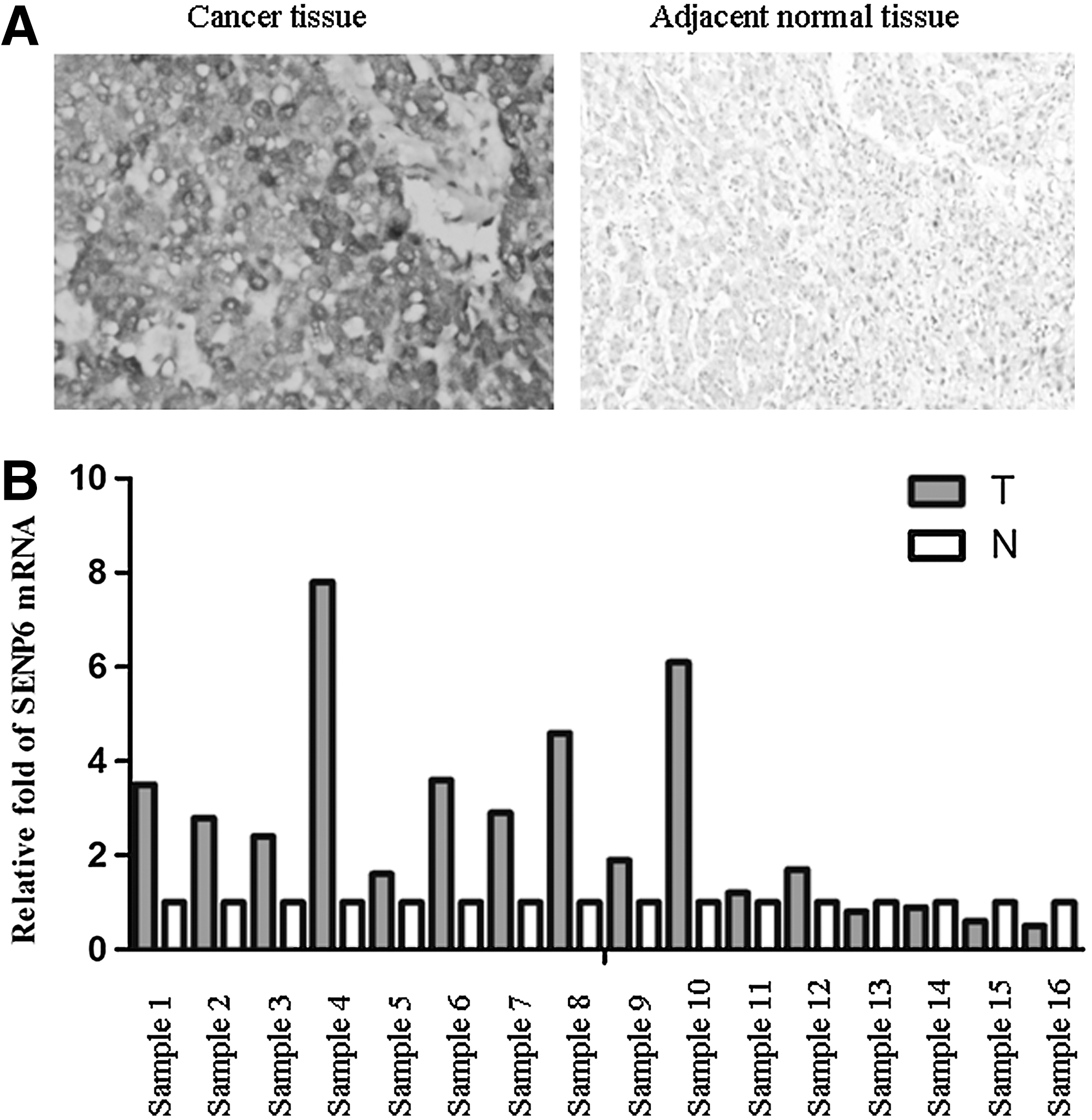
The expression of SUMO-specific protease 6 (SENP6) in human hepatocellular carcinoma tissues and adjacent normal tissues.
Inhibition of hepatocellular carcinoma cells by SENP6 silencing
To further investigate the role of SENP6 in hepatocellular carcinoma, the expression of SENP6 was silenced by lentivirus-based siRNA in a series of hepatocellular carcinoma cell lines, including HepG2, HepB3, and SK-Hep1. Compared with cells infected with a control siRNA virus vector, the mRNA expression of SENP6 was significantly silenced in the SENP6 siRNA virus-infected cells (Fig. 2A). The inhibition efficiency of SENP6 in those cells was also checked by western blot. The cells infected with the SENP6 siRNA virus showed an obviously decreased SENP6 protein expression level (Fig. 2B), consistent with the mRNA expression data. To test whether SENP6 inhibition affected the growth of these cells, the growth rates of these infected cells were compared. As shown in Figure 2C, compared with the control siRNA virus, cell proliferation was inhibited significantly in these hepatocellular carcinoma cells infected with the SENP6 siRNA virus. These data suggested a role of SENP6 in the control of proliferation of hepatocellular carcinoma cells.
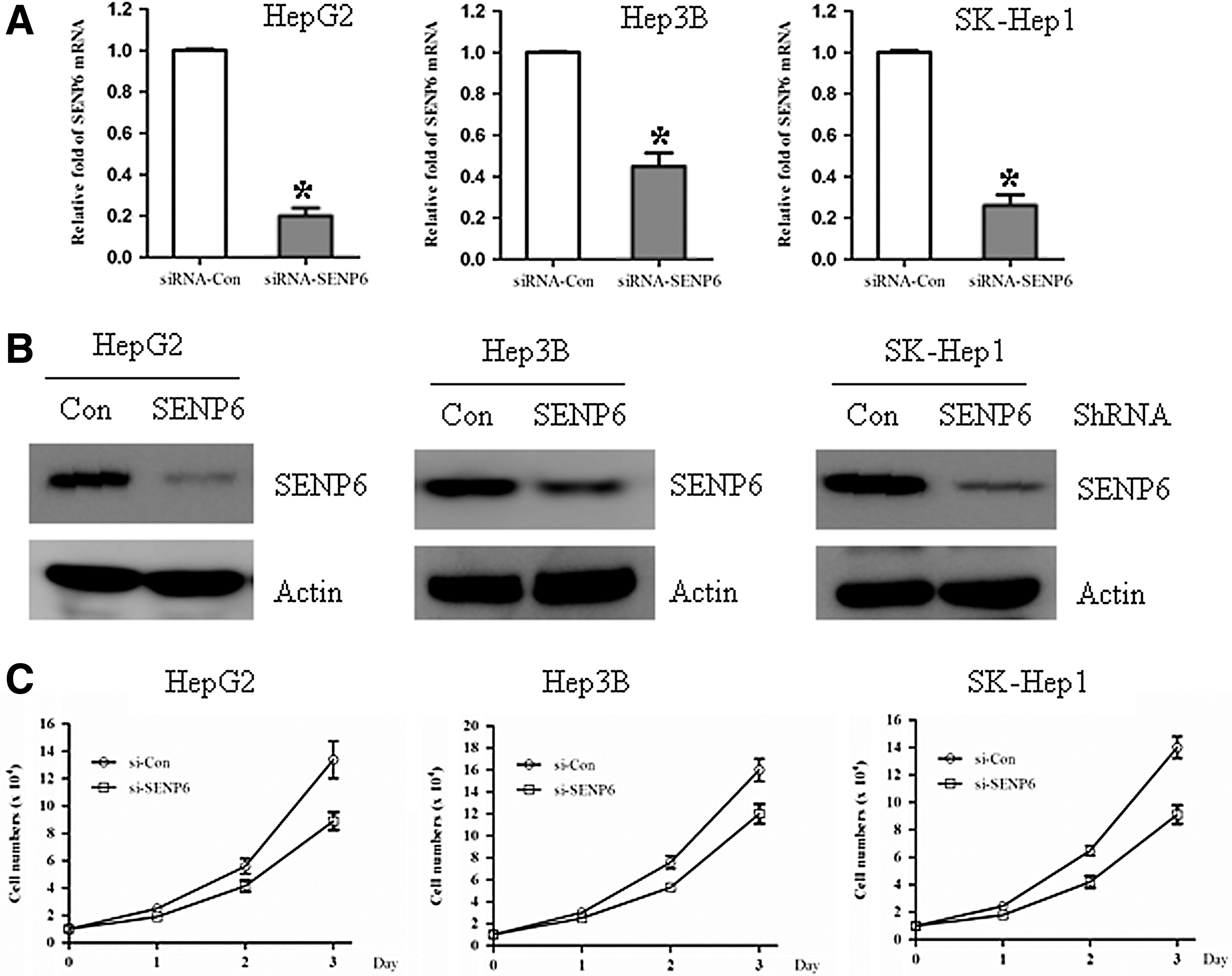
SENP6 silencing inhibited the growth of hepatocellular carcinoma cells. The hepatocellular carcinoma cell lines HepG2, Hep3B, and SK-Hep1 were infected with a con-shRNA vector and a SENP6-shRNA vector for 96 hours and then split for assays as follows.
SENP6 silencing sensitized hepatocellular carcinoma cells to radiation
SENP6 has previously been shown to have a DNA repair function. 14 Because there was a common mechanism that radiation-induced cell killing was through the production of reactive oxygen species and inhibition of DNA damage response, we determined if SENP6 silencing would affect the radiosensitivity of hepatocellular carcinoma cells. HepG2 cells infected with either the control siRNA virus or the SENP6 siRNA virus were tested in a standard clonogenic assay. As shown in Figure 3, SENP6 silencing significantly increased the radiosensitivity of HepG2 hepatocellular carcinoma cells.

SENP6 silencing sensitized hepatocellular carcinoma cells to radiation. Hepatocellular carcinoma cells, after lentivirus-based siRNA silencing, were seeded into six-well plates in duplicates. The next day, the cells were exposed to 10 Gy of radiation followed by incubation at 37°C for 8 day for colony counting. The surviving fraction was calculated and plotted after comparison with the corresponding controls (0 Gy). The sensitizing enhancement ratio was calculated as the ratio of the inactivation dose under control silencing conditions divided by the inactivation dose after SENP6 silencing.
SENP6 silencing blocks radiation-induced NF-κB activation
Ionizing radiation caused the activation of NF-κB, which in turn induced an adaptive response to confer cell resistance to ionizing radiation. 25,26 It would be reasonable to test whether SENP6 inhibition affected radiation-induced NF-κB activation. To test this hypothesis, a luciferase reporter-based transcriptional activation assay was first used to check the activity of NF-κB in the control or SENP6-silenced HepG2 cells with or without radiation. Indeed, the NF-κB transcriptional activity, as reflected by luciferase units, was approximately twofold lower at the basal level and two- to threefold lower after radiation exposure in SENP6-silenced HepG2 cells than in control-silenced cells (Fig. 3A). These data indicated that SENP6 silencing significantly blocked radiation-induced NF-κB activation.
SENP6 regulated the half-life of IκBα
To further investigate how silencing of SENP6 affected radiation-induced NF-κB activation, the protein levels of IκBα, a known inhibitor of NF-κB, in the control or SENP6-silencing HepG2 cells with or without radiation were checked. As shown in Figure 4B, the IκBα levels were much higher in SENP6-silenced cells than in the control-silenced cells at all timepoints tested. Because the decrease of IκBα is caused by enhanced protesome-dependent degradation, the higher level of IκBα in the SENP6-silenced cells may be due to an elongated protein half-life. To test this possibility, HepG2 cells were infected with the SENP6 siRNA virus, along with the control siRNA virus. About 72 hours later, the cells were exposed simultaneously to TNFα (25 ng/mL) and CHX (100 μg/mL). Indeed, SENP6 inhibition significantly increased the half-life of IκBα (Fig. 4C). Taken together, these data indicated a critical role of SENP6 in the regulation of the half-life of IκBα.
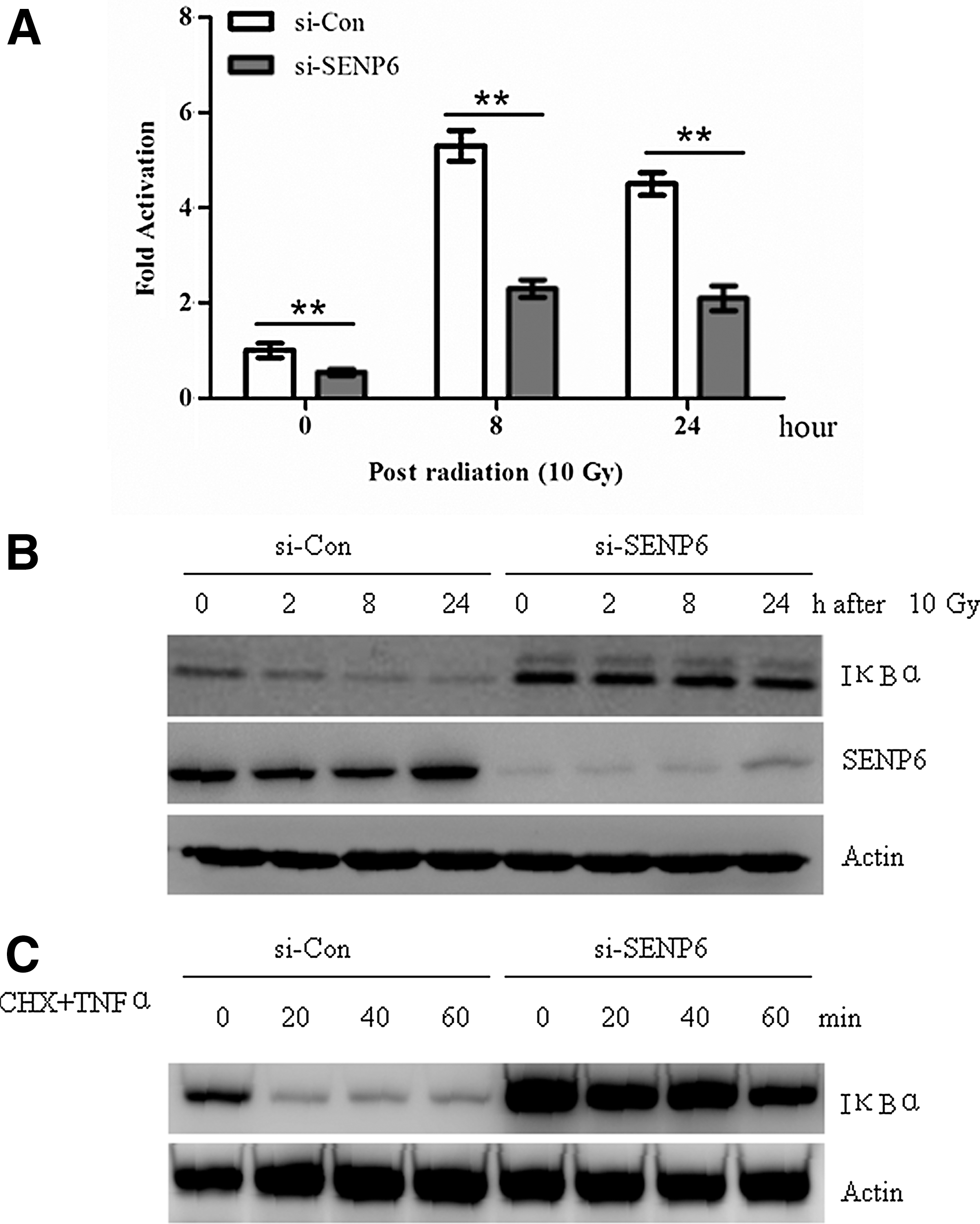
SENP6 silencing blocked radiation-induced NF-κB activation and IκBα degradation.
Discussion
In the present study, several lines of evidence were provided that SENP6 could be a promising target for hepatocellular carcinoma. First, the expression of SENP6 in clinic hepatocellular carcinoma tissues and their adjacent normal compartments was detected. SENP6 was easily detected either by immunohistochemical staining with an anti-SENP6 antibody or by real-time PCR with SENP6-specific primers in hepatocellular carcinoma tissues. However, in adjacent normal tissues, the expression of SENP6 was kept at a low level. Then, it was found that silencing of SENP6 in a series of hepatocellular carcinoma cells decreased their growth rates and increased their radiosensitivity. It was further shown that silencing of SENP6 in hepatocellular carcinoma cells blocked radiation-induced NF-κB activation by preventing the degradation of IκBα, which is a well-known inhibitor for NF-κB.
Radioresistance was a major obstacle for effective cancer treatment. Particularly, in cases of hepatocellular carcinoma, radiotherapy was very ineffective due to the extreme radioresistance of cancer cells, contributing to the poor patient survival rate. Here, it was shown that SENP6 may contribute to the radioresistance of hepatocellular carcinoma, because it was overexpressed in hepatocellular carcinoma, and SENP6 inhibition rendered that these cancer cells were sensitive to radiation.
The most attracting role of SENP6 was silencing of SENP6-blocked radiation-induced NF-κB activation. The NF-κB pathway was a driving force for the development of numerous cancers and provided cancer cells with a radioresistance ability. 27 The exact role of SENP6 in the regulation of the NF-κB pathway, especially how SENP6 regulated the stability of IκBα, should be elucidated by additional investigations. The future challenges will be to identify specific inhibitors of SENP6 and to develop them as a novel class of anticancer agents and radiosensitizers combined with radiation to treat hepatocellular carcinoma.
Footnotes
Disclosure Statement
None of the authors have any potential conflicts of interest.