
Abstract
Chemoresistance remains a major clinical obstacle to successful cancer treatment and brings about poor prognosis of the patients, yet the underlying mechanisms have not been entirely understood. MicroRNAs (miRNAs) are a new class of small noncoding RNAs that may play an essential role for regulation of programmed cell death, which consists of apoptosis and autophagy. Autophagy refers to an evolutionarily conserved catabolic process in which, a cell degrades long-lived proteins and damaged organelles. Recently, increasing evidence indicates that autophagy is associated with multiple cancer-related pathways, including resistance to chemotherapeutics. Moreover, manipulation of miRNA expression levels may increase cell sensitivity to cytotoxic drugs through targeting the autophagic signaling pathway. In this review, we summarized the recent findings concerning miRNAs involved in autophagy, mainly focused on the mechanism of miRNA modulation at different autophagic stages, the crucial role of miRNAs in the interconnection between autophagy and apoptosis, and the potential of miRNAs to overcome chemoresistance by targeting autophagic pathways.
Introduction
It is well-known that chemotherapy is one of the main adjuvant cancer treatments to postpone the relapse of cancer. However, a major clinical obstacle to the successful treatment continues to be the intrinsic or required resistance toward the therapy, and the underlying mechanisms have not been entirely understood. Since the high-level autophagy in tumor cells following anticancer treatment is considered to be a survival mechanism, therapeutic targeting of the autophagic signaling pathway might represent a novel molecular avenue to reduce the occurrence of chemoresistance. 1
Autophagy, also called macroautophagy, is a lysosome-mediated intracellular self-catabolic degradation process by which, cells may remove their damaged organelles, as well as unfolded and aggregated proteins so as to maintain cellular homeostasis. 2 Of note, the role autophagy played in oncogenesis is double sided and context dependent. 3 In a tumor microenvironment, autophagy can serve as a means of temporary survival in response to metabolic stress; meanwhile, once the cellular stress results in continuous or progressive autophagy, cell death would follow. Moreover, emerging evidence has affirmed autophagic features in cells treated with chemotherapeutic agents. 3 The question that the high level of autophagy induced by cytotoxic drugs should be regarded as a direct cell death execution pathway or a garbage disposal mechanism whereby cells preserve their viability for long-time survival still needs clarification. 4 Recently, work on one group of endogenous noncoding RNAs, miRNAs, has expanded our recognition of the regulation of autophagy. 5
MicroRNAs (miRNAs) are a new class of small noncoding RNAs that have been highly conserved during evolution and have been recently estimated to regulate gene expression at post-transcriptional and translational levels. 6 Dysregulation of miRNA expression would lead to many human diseases, including cancers. 7 Moreover, an increasing number of researches have hinted that manipulation of miRNA expression levels may increase cell sensitivity to cytotoxic drugs through targeting autophagic signaling pathways. 8 –11 Intriguingly, a group of studies have highlighted the pivotal regulatory roles of miRNAs in apoptotic signaling pathways, with substantial evidence manifesting that suppression of apoptosis induces autophagy, and autophagy inhibition causes apoptosis. 12 Here we summarized recent advances of miRNA regulation in autophagy and apoptosis and tried to make discussion and exploration simply for the association of miRNA-mediated autophagic signaling networks with cancer chemoresistance.
Both Oncogenic and Tumor Suppressive miRNAs Regulate Autophagy
Recently, accumulating work on miRNAs, consisting of oncogenic and tumor suppressive ones, show their capabilities in modulating some members of autophagy genes (Atgs) and their regulators at different autophagic stages, including induction, vesicle nucleation, and vesicle elongation and completion, which produce the mature autophagosome that then fuses with a lysosome for degrading and recycling (Fig. 1).
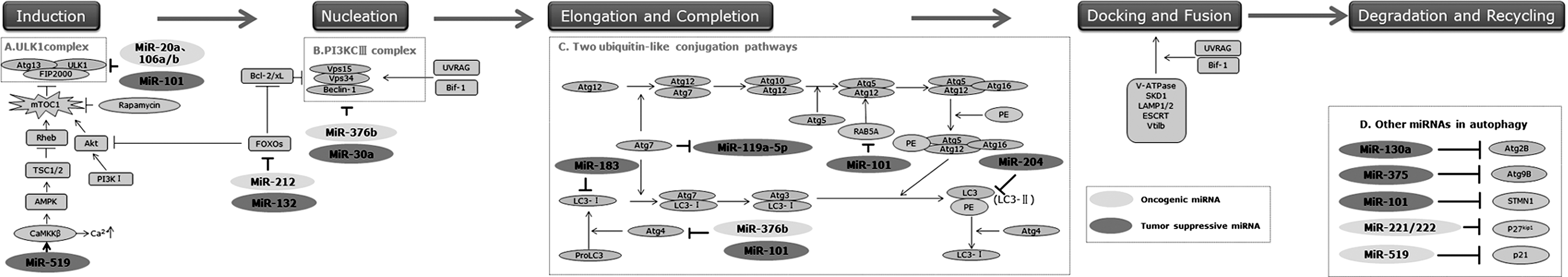
MicroRNAs (miRNAs) modulate different autophagic stages. MicroRNAs, consisting of oncogenic and tumor suppressive ones, can modulate different autophagic stages, mainly focus on several signaling pathways:
First, autophagy induction is initiated by activation of the Unc-51-like kinase (ULK1) complex, which could be inhibited by mammalian target of rapamycin complex 1 (mTORC1) that serves as a main regulator for autophagy (Fig. 1). MiRNAs could discourage the autophagosome nucleation through targeting mTORC1, and then disrupting the binding of the ULK1 complex via the phosphorylation of two of its components, ULK1 and Atg13. 13 Both oncogenic miRNAs, such as miR-20a and miR-106a/b, 14,15 and tumor suppressive miRNAs, such as miR-101, 16 could regulate autophagy via targeting the ULK1 complex. Furthermore, the fact that miR-106a directly blocked ULK1 activation in NB4 acute myeloid leukemia cells also identified ULK1 as a potential tumor suppressor. 15 Additionally, the intracellular energy-sensing pathway targets mTORC1 by modulating the activity of adenosine monophosphate-activated protein kinase (AMPK). 17 There has been a rising recognition that calcium mobilization excited autophagy via activating the calcium/calmodulin kinase (CamKK)β, one upstream kinase of AMPK. 18 Recent data showed that tumor suppressive miRNA, miR-519, modulated intracellular calcium levels, especially via the changes in the abundance of calcium release-activated calcium modulator 1(ORA I 1) and ATPase 2C1 (ATP2C1), two proteins that are essential for maintaining cytosolic calcium homeostasis, and then directly leading to the promotion of autophagic cell death. 19 The mTOR-independent pathway of autophagy regulation also existed, but less well understood. For instance, induction of autophagy occurs via FoxO-dependent transcription of autophagy-related genes, such as microtubule-associated protein 1 light chain 3B (MAPLC3B, also named LC3B) and BCL-2/adenovirus E1B protein-interacting protein 3 (BNIP3). 20 Both miR-212 and miR-132 have been found to negatively modulate the expression of the FoxO3 transcription factor, a member of the FoxO transcription factor family. 21
Second, the vesicle nucleation is mediated by activation of the Beclin-1-class III phosphatidylinositol 3-kinase (PI3KCIII, an ortholog of Vps14)-Vps15 core complex and other proteins 22 (Fig. 1). In this step, miR-30a with a tumor suppressive role has been demonstrated to negatively regulate the expression of Beclin-1. Interestingly, another oncogenetic miRNA, miR-376b, was recently shown to attenuate rapamycin-induced autophagy by directly downregulating the translational level of Beclin-1 in MCF-7 and Hun-7 cells. 23
Third, the vesicle elongation and completion process is mediated by two ubiquitin-like conjugation pathways, which mainly involve two ubiquitin-like proteins, Atg12 and LC3 (microtubule-associated protein 1 light chain 3, a mammalian ortholog of Atg8), respectively 24 (Fig. 1). After the autophagosome expansion completion, LC3 detaches from phosphatidylethanolamine under the cooperation of Atg4, and then releases to cytosol. 25 Downregulation of miR-183 and miR-204 could boost the autophagy process by promoting the activation of LC3B. 26,27 Atg4-Atg8 conjunction is a vital touch in the autophasosome biogenesis pathway, and Atg4 has been considered to be the main regulator of LC3 in mammalian cells. As an example, autophagy can be potently inhibited by miR-101 and miR-376b, via negatively regulating the expression of Atg4C and Atg4D, respectively. 16,23 In addition, other two bona fide targets of miR-101 are Stathmin 1(STMN1) and RAB GTPase 5A (RAB5A). 16 STMN1 destabilized microtubules and regulated cell cycle, 28 while RAB5A was indicated to regulate Atg5-Atg12 conjugation in the autophagosome completion pathway. 29
Fourth, autolysosome maturation is promoted by docking and fusion of autophagosome with endolysosomal compartments, eventually leading to the breakdown of autophagosomal contents. 30 This process requires the cooperation of microtubules and endolysosomal molecules, containing small GTPase Rab7, lysosomal-associated proteins 1 and 2 (LAMP1 and LAMP2), AAA ATPase SKD1, and other proteins. In the last step of degradation and recycling, the acidic hydrolases impel the digestion of autophagosomal cargoes, and then nutrient and energy are recycled. So far, there is little report affirmed miRNA function in the last two steps, yet.
Additionally, Xu et al. also found that downregulation of miR-199a-5p enhanced autophagy activation via targeting Atg7, which increased drug resistance in human hepatocellular carcinoma cells. 10 Again, miR-130a and miR-375 have been identified as strong inhibitors of basal and induced autophagy by downregulating Atg2B and Atg9B, respectively. 31,32 Some miRNAs can indirectly impact on the autophagy process through cell cycle modulation. MiR-221/222 has been confirmed to inhibit the cell cycle inhibitor p27 kip1, which acted as a downstream modulator of PI3K I/Akt, resulting in autophagic cell death in the HER2/neu-positive primary human breast carcinoma MCF-7 cell. 33 Another similar example is p21, known as the cyclin-dependent kinase inhibitor, whose level can be elevated by miR-519-triggered calcium upregulation, which ultimately repressed tumor growth at least in part by triggering autophagy. 19
As aforementioned, although both oncogenic and tumor suppressive miRNAs might modulate the autophagy process, the underlying mechanisms remain largely unknown. Recently, scholars have put forward one hypothesis that overexpression of oncogenic miRNAs may bring about tumorigenesis by inhibiting tumor suppressive gene expression or genes that control cell differentiation or apoptosis; likewise, underexpression of tumor suppressor miRNAs may bring about incorrect expression of oncogenes or genes that inhibit cell differentiation or apoptosis. 34 It is consistent with what has been found in the miRNA-mediated autophagic signaling pathway. Some tumor suppressors, such as the ULK1 complex, Beclin-1, and Atg4, can be downregulated by oncogenic miRNAs; similarly, oncogene such as mTORC1 can be downregulated by tumor suppressive miRNAs. However, Beclin-1 and Atg4 are regulated by both oncogenic and tumor suppressive miRNAs. Thus, it can be seen that miRNA-mediated autophagy is not at the level of a single gene product, but the entire network.
The Emerging Roles of miRNAs in Cross Talk Between Autophagy and Apoptosis
At first, one notion should be made clear that both autophagy and apoptosis function in the cell growth, survival, development, and death. Due to this point, these two pathways might be triggered by a common upstream signal, which implies at least one shared molecular switch, therefore, resulting in the combination or mutual exclusion of them. Remarkably, some confirmed targets of autophagy miRNAs are involved in the main interconnection routes linking autophagy and apoptosis (Fig. 2).
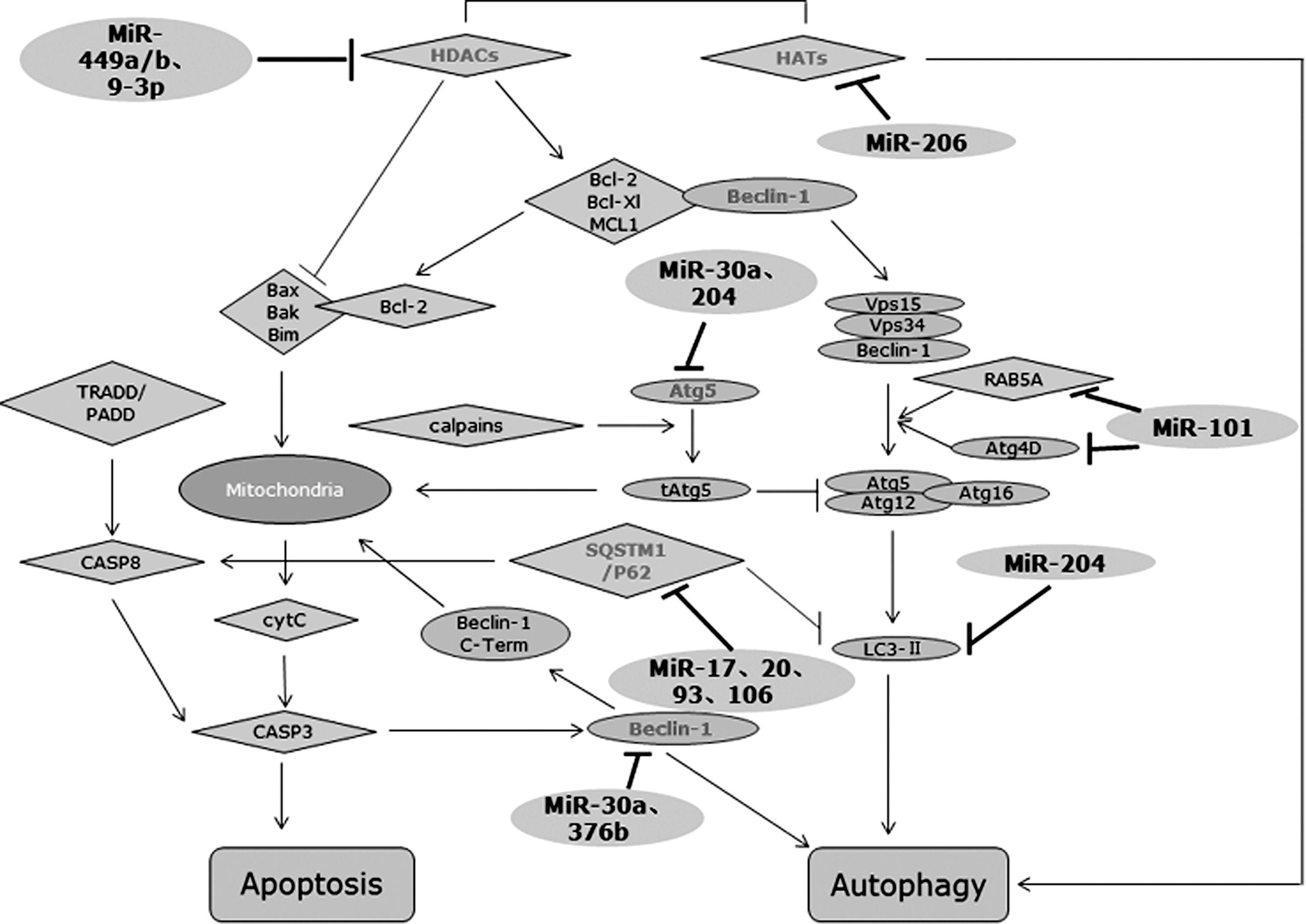
The emerging roles miRNAs played in cross talk between autophagy and apoptosis. In addition to targeting autophagy-associated proteins such as Atg4D, RAB5A, and LC3-II, miRNAs can also mediate the cross talk between autophagy and apoptosis via targeting the shared switch molecules such as Beclin-1, Atg5, and SQSTM1, and via changing the balance of nucleosomal histone acetylation.
miRNAs and Beclin1/Bcl-2 complex
Beclin-1, which is well documented to arrest autophagy through the Beclin-1 BH3 domain with the antiapoptotic family consisting of B-cell lymphoma/leukemia-2(Bcl-2), B-cell lymphoma-extra large (Bcl-xl), and myeloid cell leukemia sequence-1 (MCL-1), certainly plays a major role in this cross regulation. 35 –37 Take Bcl-2, for example, under nutrient excess conditions, Beclin-1 and Bcl-2 bind to each other, and autophagy is not necessary at this moment; conversely, c-Jun N-terminal protein kinase-1 (JNK-1)-mediated Bcl-2 phosphorylation leads to its dissociation from Beclin-1, thus simultaneously inhibiting apoptosis and promoting autophagy (Fig. 2). Several findings have demonstrated that the cytoplasmic level of Beclin-1 might be decreased by overexpression of miR-30a in both solid cancer cells and human chronic myeloid leukemia (CML) cells. 11,38 Moreover, the latter study further indicated that induction of autophagy via downregulating miR-30a restrained imatinib-induced intrinsic apoptosis, whereas the upregulated miR-30a increased imatinib-induced cytotxicity. 38
miRNAs and Atg5
The existence in the interplay between autophagy and apoptosis is also sustained by the double roles of Atg5, which not only increased the autophagic activity, but also increased the likelihood to undergo apoptosis. It was shown that cleavage of Atg5 by calpain during apoptosis allows Atg5 translocation from the cytosol to mitochondria, where it associated with Bcl-xl and cytochrome c release, and finally triggered caspase activation and apoptosis (Fig. 2). 39 MiR-204, which has an antiapoptosis effect, may also inhibit cardiomyocyte autophagy. 40 Later, Atg5, from another report referred to renal clear cell carcinoma, has been found to be the downstream target of miR-204. 27 However, downregulation of Atg5 expression might also trigger cells from autophagy to apoptosis. Upregulation of miR-30a, which, in addition to Atg5 inhibition, promoted imatinib-induced apoptosis, and as a result, restored the sensitivity to imatinib in CML cells. 38 This apparent discrepancy might arouse a problem as to how Atg5 can cope with the relationship between autophagy and apoptosis. It is possible that the level of Atg5 expression influences cells to turn to one or another. Overexpression of Atg5 contributes to cleavage of itself, thus inducing autophagy as well as apoptosis; in contrast, after downregulating the expression of Atg5 via various upstream modulators, autophagy might be suppressed due to lower levels of Atg5 expression, triggering cells toward apoptosis through other signaling pathways. Only the level of Atg5 expression fluctuating in a certain range can prompt cells to enter autophagy rather than apoptosis. As the upstream modulators of Atg5, miRNAs are likely to assist in changing its expression level, thereby guiding cancer cells to one another and rendering cells exhibit sensibility or resistance to chemotherapy as a result.
miRNAs and SQSTM1/P62
Sequestosome1/p62(SQSTM1/P62), one multidomain protein acting as a signal, which in addition to negatively regulating the degradation of the autophagic protein LC3 via binding to it through a region termed LC3-interacting region, could also modulate the polyubiquitination and aggregation of caspase-8 that is required to trigger the apoptotic pathway (Fig. 2). 41 SQSTM1 was identified as a main target for miR-17, 20, 93, and 106, which shared the identical AAGUGC seed region and specific target. 42 Upregulation of these AAGUGC seed-containing miRNAs contributes to the elimination of SQSTM1, that is to say, acceleration of autophagy, but no experimental data show its influence on the apoptotic pathway. Thus, the potential mechanism of miRNA-mediated SQSTM1 underlying the cross talk between autophagy and apoptosis requires intensive study.
miRNAs and histone acetylation
The balance of nucleosomal histone acetylation maintained by a tight regulation of the level of histone deacetylases (HDACs) and histone acetyl transferases (Fig. 2), impacts a broad repertoire of physiological processes, including autophagy and apoptosis. 43 This balance is disrupted in many cancer cells, with an increased expression of HDACs. HDAC inhibitors, such as sirtinol, have been demonstrated to prompt tumor cells to enter apoptosis via downregulating prosurvival proteins containing Bcl-2 and Bcl-xl, and via upregulating proapoptotic proteins containing Bim, Bak, and Bax, as well as to enter autophagy. 44,45 It is noteworthy that pretreatment with the autophagy inhibitor such as 3-methyladenine might increase the sirtionl-induced cell cytotxicity, which is related to blocking autophagic cell death and increasing apoptotic cell death. 44 On the basis of the properties, autophagy induced by HDAC inhibitors may be just a mechanism of resistance rather than cell death. MiR-449a/b has been showed to regulate the level of histone acetylation via suppressing substantial expression of HDAC1 in lung cancer. 46 Nevertheless, HDACs have also been found to mediate the epigenetic silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia cells, and elevate the levels of MCL-1. 47 Thereby, the point that HDAC is upregulation of miRNAs as the cause or effect of the event has not been determined. An obvious explanation is cell-type specificity. Alternatively, cells may choose different routes to link HDAC with modulation of miRNAs. Besides, another latest investigation suggested increased expression of miR-206 and reduced expression of miR-9-3p in primary waldenstrom macroglobulinemia cells deregulated histone acetyation and brought about autophagy-dependent cell toxicity, 48 but the mechanism histone actylation affecting autophagy remained largely unknown. An opposite case is that suppression of HDAC attenuates cardiac hypertrophy via autophagy inhibition. 49 Therefore, future investigations are needed to elaborate the potential molecular mechanism, for further assisting in explaining this discrepancy.
Potential of miRNAs to Overcome Chemoresistance
Given the fact of the abnormal expression of specific miRNAs in drug-resistant tumor cells, sensitivity to chemotherapy can be recovered by regulating these miRNA expressions with various mechanisms that are still not entirely understood. As stated above, the roles that miRNAs played in the autophagy process and in the cross talk between autophagy and apoptosis signals, reveal a bran-new resistance mechanism related to dysregulation of miRNA expression.
In this working model, depending on multitarget characteristics, miRNAs can act as molecular on-off switches to completely turn off a cellular process, with or without turn on another cellular process. As an example of the autophagy process, one single miRNA can concurrently target multiple autophagy genes that control different stages, for instance, miR-101 repressed three autophagy-associated genes, Atg4D, STMN1, and RAB5A. 16 Alternatively, several miRNAs may target the same autophagy gene or autophagic stage in a cooperative manner. Both miR-376b and miR-30a downregulated the translational levels of Beclin-1, thus resulting in the dissociation of the Beclin-1-PI3KCIII-Vps15 core complex, as well as the protein–protein interactions with proapoptotic proteins, Bcl-2 and Bcl-xl. 38,23 The inhibition of autophagy and the promotion of apoptosis regulated by miRNAs strengthen the sterilization of cytotoxic drugs to cancer cells. As mentioned, downregulation of miR-30a, as well as miR-199a-5p, increased the tolerance of tumor cells to cytotoxic drugs by activating autophagy and simultaneously suppressing apoptosis. 10,11 All of these findings provide a reasonable probability for the development of therapeutic approaches based on miRNA-modulating agents in combination with conventional cytotoxic drugs via targeting the autophagic signaling pathway.
Conclusion and Perspective
Cancer is a multistep human disease that is related to a series of complex biological processes, certainly encompassing autophagy. Currently, numerous studies have been conducted to determine the molecular mechanism of autophagy, to explicit the role that autophagy played in cancer initiation and progression, and to elucidate the significance of targeting the autophagic signaling pathway to anticancer therapeutics. In this review, the recent findings concerning miRNAs involved in autophagy have been summarized, mainly focused on the mechanisms of miRNA modulation in different autophagic stages, as well as the crucial role of miRNAs in the interconnection between autophagy and apoptosis. Such efforts are imperative to better understanding of miRNA-mediated autophagy in cancer and to apply in appropriate therapies targeting this small class of molecules to reduce the occurrence of chemoresistance.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81071806 and 81172106) and the National Natural Science Foundation of Jiangsu Province (Grant No. BK.2012371).
Disclosure Statement
No competing financial interests exist.