
Abstract
In this study, we investigated the potential anticancer effect and mechanisms of action of a methanol extract from Aristolochia debilis Sieb. et Zucc (A. debilis) stems on human colon cancer cells (HT-29). A. debilis inhibited proliferation of HT-29 cells in dose-dependent and time-dependent manners as detected by the 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan assay. A cell cycle analysis was performed via propidium iodide staining and flow cytometry. The A. debilis extract affected cell cycle progression by inducing sub-G1 arrest. The reactive oxygen species (ROS) level was determined by dichlorofluorescin diacetate. A decrease in the mitochondrial membrane potential and inhibition of manganese superoxide dismutase in combination with the accumulation of ROS induced apoptosis. Reverse transcription-polymerase chain reaction and Western blot analyses were used to determine changes in the expression of mitochondria-dependent apoptosis markers (Bax and Bcl-2). Upregulation of Bax and corresponding downregulation of Bcl-2 expression as well as ROS production may be the critical mechanism through which A. debilis induced apoptosis in HT-29 cells.
Introduction
Aristolochia species contain aristolochic acid (AA) and nitrophenanthrene carboxylic acids, which have been used in Traditional Chinese Medicine as diuretics and analgesics, respectively. Nevertheless, an overdose of AA is nephrotoxic and genotoxic. 1 The U.S. Food and Drug Administration has reported that Aristolochia products may have potential carcinogenic properties. 2 Therefore, most governments have forbidden the use of Aristolochia species as a medicinal herb or as an ingredient in medicinal products. Based on AA poisoning, researchers began to investigate safe and toxic dosages. 3,4 A low dose of Aristolochia debilis Sieb. et Zucc (A. debilis) is safe and is not renal toxic. 4 Furthermore, the Aristolochia family has antiproliferative effects on the human MCF-7 breast cancer cell line. 5
The U.S. International Cancer Center reported that deaths from colon and rectal cancer account for about 10% of all deaths from cancer. 6 Cancerous cells continuously produce reactive oxygen species (ROS) during the cell cycle, which increases oxidative stress. 7,8 The increased quantity of ROS in cancer cells may stimulate cellular proliferation, promote mutations and genetic instability, and alter cellular sensitivity to anticancer agents. 9 Superoxide dismutase (SOD), a key antioxidant enzyme, effectively regulates antioxidant defense to maintain appropriate intracellular ROS levels and prevent oxidative damage. 8,9
Apoptosis, a gene-directed cell death process, is characterized by cell shrinkage, membrane blebbing, chromatin condensation, and nuclear fragmentation. 10,11 Apoptosis is induced via two main routes involving either mitochondria (the intrinsic pathway) or activation of the death receptors (the extrinsic pathway). Apoptosis is important for controlling cell numbers and proliferation and is the major control mechanism for inducing no repaired DNA damage cell self destruction. 12 However, cancer cells do not undergo apoptosis. Thus, drugs and agents that can restore the normal apoptotic pathway may have potential for treating cancers. Therefore, in the present study, we investigated the potential molecular mechanism of apoptosis induced by an A. debilis extract in the HT-29 human colon cancer cell line.
Materials and Methods
Sample preparation
A. debilis stems were collected from native trees (Yunnan, China). The fresh stems were air-dried and then ground to a fine powder. Fifty grams of powder was extracted twice with 1 L of methanol. The extract was filtered by vacuum (100 mm; Whatman, Maidstone, United Kingdom), and the solvent was evaporated under reduced pressure using a vacuum rotary evaporator (CCA-1110; Eyela, Tokyo, Japan). The dried extract was stored at −20°C for subsequent analysis.
Cell lines and cell culture
Embryonic kidney cell (HEK293) and human colon cancer cell (HT-29) lines were purchased from the Korean Cell Line Bank (Seoul, Korea) and grown in the Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute medium 1640 (RPMI 1640), respectively, supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured in a humidified atmosphere and incubated at 37°C in 5% CO2.
Cytotoxicity test
The HEK293 and HT-29 cells were seeded in 96-well plates (1×105 cells/well) and incubated in the DMEM and RPMI 1640, respectively, at 37°C in 5% CO2 for 24 hours. The HEK293 cells were pretreated with 50, 100, 200, 400, and 800 μg/mL A. debilis extract and the HT-29 cells were incubated with 50, 100, and 200 μg/mL A. debilis extract for 24 hours. In addition, another group of HT-29 cells was incubated with/without N-acetyl-L-cysteine (NAC, 5 mM) and 200 μg/mL A. debilis extract for 6, 12, and 24 hours. After the incubation, a 20-μL aliquot of 2 mg/mL MTT solution was added to each well for 4 hours. Then, 200 μL of dimethyl sulfoxide was added to stop the reaction, after discarding the supernatant. The absorbance was measured at 550 nm using an enzyme-linked immunosorbent assay plate reader (Bio-Tek, Winooski, VT).
Cell cycle analysis
HT-29 cells were seeded in six-well plates (1×106 cells/well) for 24 hours. Then, the cells were incubated with 50, 100, and 200 μg/mL A. debilis extract for 24 hours. The cells were collected and washed twice with phosphate-buffered saline (PBS). Each group (1×106) was fixed in 1 mL fixative at −20°C for 2 hours. After decanting the fixative, the cell pellet was suspended in 0.5 mL of the propidium iodide staining solution. After a 30-minute incubation at room temperature in the dark, the cells were analyzed by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ).
Measurement of ROS
The level of intracellular ROS was measured by the change in fluorescence resulting from oxidation of dichlorofluorescin diacetate (DCFH-DA). Cells were seeded in six-well plates (1×106 cells/well) for 24 hours. After the incubation, the cells were treated with different concentrations of the A. debilis extract with/without NAC for 6, 12, and 24 hours. Then, the cells were collected, washed with PBS, and treated with 20 μM DCFH-DA for 30 minutes in the dark. Fluorescence was measured on a FACSCaliber flow cytometer (Becton Dickinson) with a 485 nm excitation filter and a 535 nm emission filter, and data were analyzed with the Cell Quest program.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) was measured by the change in fluorescence from 3,3′-dihexyloxacarbocyanine iodide (DiOC6). Briefly, cells were seeded in six-well plates (1×106 cells/well) for 24 hours. Following a 24-hour treatment with different concentrations of the A. debilis extract, the cells were washed with PBS and incubated with 20 nM DiOC6 for 20 minutes. The cells were collected and washed again with PBS. Fluorescence was measured with a FACSCaliber flow cytometer with a 484 nm excitation filter and a 501 nm emission filter, and data were analyzed with the Cell Quest program.
RNA isolation and reverse transcription-polymerase chain reaction analysis
Total RNA was isolated from A. debilis-treated cells using a Trizol RNA Isolation kit (Invitrogen, Carlsbad, CA) and stored at −80°C for subsequent analysis. One microgram of RNA was reverse transcribed into cDNA and subsequently used as the template for reverse transcription-polymerase chain reaction (RT-PCR) amplification. The primers used were SOD2, 5′-GCTAACATTCTCCCAGTTGA-3′ and 5′-GTGACTTTGGGTCTTTTGAG-3′; Bax, 5′-TCCACCAAGAAGCTGAGCGA-3′ and 5′-GTCCAGCCCATGATGGTTCT-3′; Bcl-2, 5′-TGTGGCCTTCTTTGAGTTCG-3′ and 5′-TCACTTGTGGCTCAGATAGG-3′; β-actin, 5′-TCACCCTGAAGTACCCCATC-3′ and 5′-CCATCTCTTGCTGCAAGTCC-3′. The conditions for SOD2 and Bcl-2 were 94°C for 5 minutes, and amplification was followed by 30 cycles of denaturing at 94°C for 30 seconds, annealing at 60°C for 30 seconds, primer extension at 72°C for 45 seconds, and a final extension at 72°C for 7 minutes. The PCR conditions for Bax and β-actin were 94°C for 5 minutes, and the amplification was followed by 25 cycles of denaturing at 94°C for 30 seconds, annealing at 55°C for 30 seconds, primer extension at 72°C for 45 seconds, and a final extension at 72°C for 7 minutes. The PCR product was analyzed on a 1% agarose gel, and DNA bands were visualized by ethidium bromide staining and a Mini BIS Image Analysis System (DNR Bio-Imaging Systems Ltd., Kiryat Anavim, Israel).
Western blot analysis
Cells (1×106 cells) were incubated in 6-well plates for 24 hours. After the incubation, the cells were treated with 50, 100, and 200 μg/mL A. debilis extract for 24 hours. Then, the cells were harvested, washed with cold PBS, and incubated in a lysis buffer (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 2 mM EGTA, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM dithiothreitol, 1% Triton X-100, 10% glycerol, 10 mg/mL leupeptin, 10 mg/mL aprotinin, 10 mg/mL pepstatin, 1 mM benzimidine, and 2 mM phenylmethane sulfonylfluoride) for 30 minutes at 4°C. Cell and tissue lysates were centrifuged for 10 minutes at 13,000 g. Supernatants were collected, and the total protein concentration was determined using the Bradford assay. A 30 g quantity of proteins was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene difluoride membrane (Bio-RadHercules, CA). Nonspecific binding was blocked with 2% bovine serum albumin in Tris-buffered saline (4 mM Tris base TBS and 100 mM NaCl) containing 0.2% Tween 20 (TTBS) and incubated for 1 hour with the indicated primary antibodies and for 1 hour with the HRP-conjugated secondary antibody, and then washed three times with the TTBS buffer. Blots were developed with the Mini BIS Image Analysis System and were quantified with the Quantity One image analysis software program.
Statistical analysis
All tests were carried out independently in triplicate. Data are expressed as mean±standard derivation. Statistical significance was evaluated with analysis of variance using SPSS 16.0 (SPSS, Inc., Chicago, IL). Results were considered significant when p<0.05.
Results
Cytotoxicity and antiproliferative effects of the A. debilis extract
An overdose of the Aristolochia plant could lead to serious renal failure. 13,14 Therefore, we estimated the toxicity of A. debilis in normal HEK293 cells and found that the extract was slightly toxic to these cells at concentrations ≤200 μg/mL (Fig. 1A). The antiproliferative effect of A. debilis on HT-29 cells (Fig. 1B) was dose dependent as determined by the MTT assay. Approximately, 68.4, 51.8, and 32.5% HT-29 cell viability was observed after treatment with 50, 100, or 200 μg/mL A. debilis, respectively.
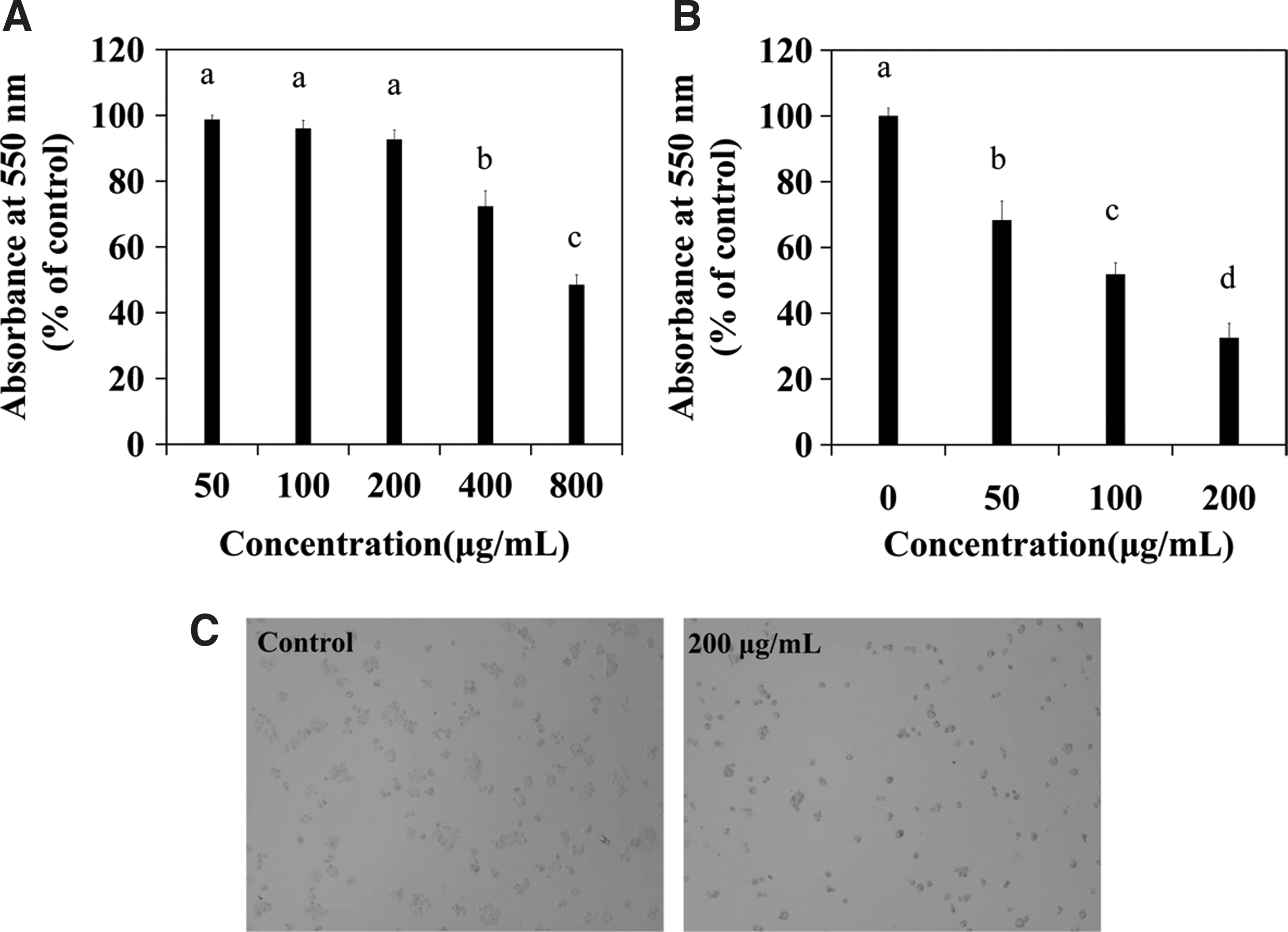
Cytotoxicity and antiproliferative effect of A. debilis.
Furthermore, morphological changes in A. debilis-stimulated HT-29 cells were observed directly by light microscopy (Fig. 1C). Cultures not treated with the A. debilis extract contained tight colonies, and the individual cell boundaries could not be delineated. In contrast, cultures treated with various concentrations of A. debilis showed wider intercellular spaces, and the cells were round. Individual cell boundaries were clearly identified in the 200 μg/mL A. debilis-treated culture.
A. debilis extract induces sub-G1 cell cycle arrest in HT-29 cells
As the A. debilis extract decreased HT-29 cell proliferation, we further investigated whether this effect was due to cell cycle arrest. The flow cytometry analysis revealed that 29.38%, 40.38%, and 42.82% of the cells were arrested at the sub-G1 phase after being incubated with 50, 100, or 200 μg/mL A. debilis, respectively (Fig. 2B). In the untreated group, 74.96% of HT-29 cells were in the G0/G1 phase; however, after treating the cells with increasing concentrations of the A. debilis extract, the percentage of cells in G0/G1 declined sharply. The cell population at the S and G2/M phases did not change.
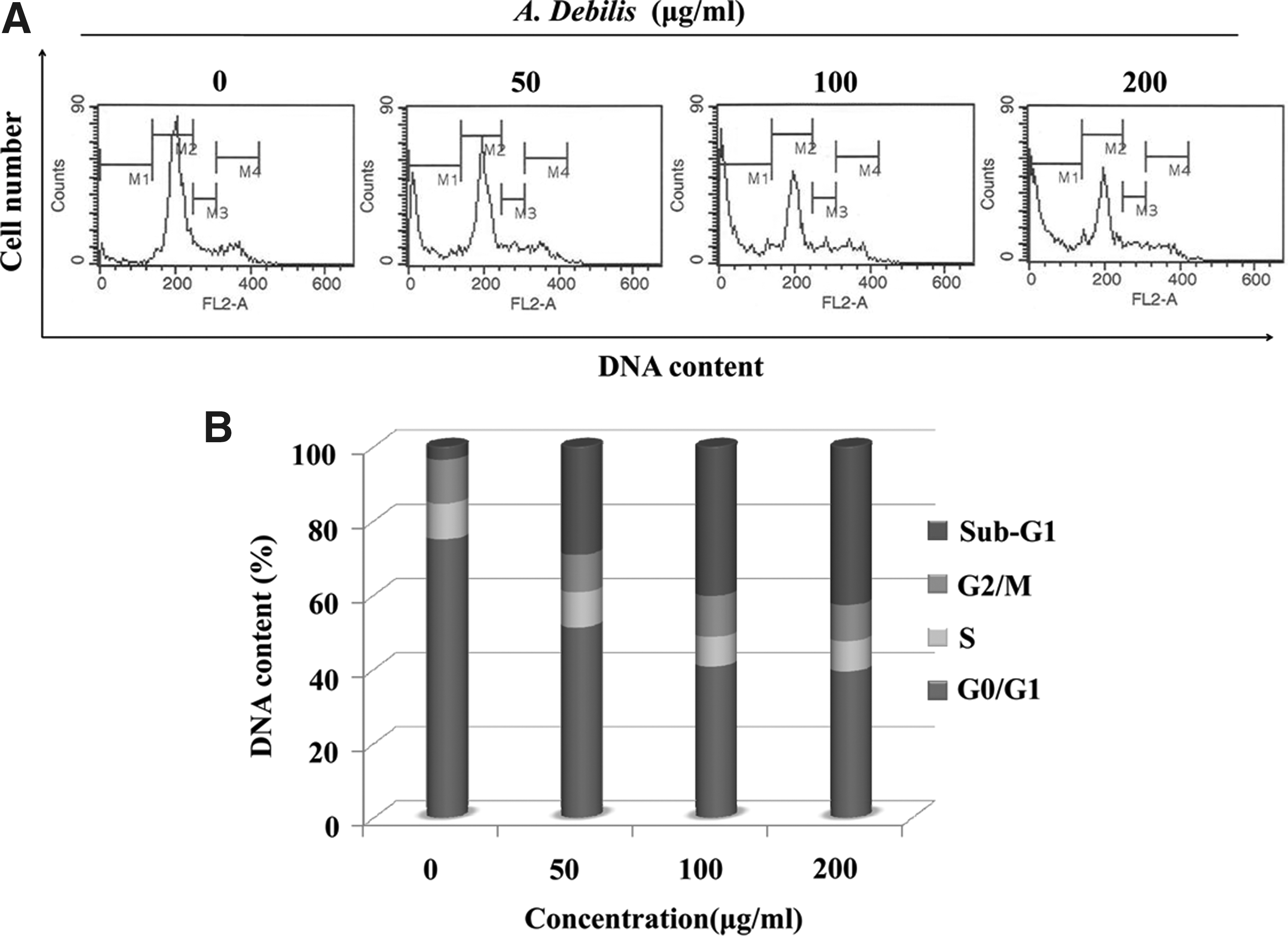
Cell cycle analysis on human colon cancer cells (HT-29) incubated with varying concentrations of A. debilis for 24 hours.
A. debilis extract induces cell death by generating ROS and decreasing the MMP
To investigate whether A. debilis-induced cell death was related to the generation of ROS, the ROS level in HT-29 cells was measured using DCFH-DA. As shown in Figure 3A, the ROS level in A. debilis-stimulated HT-29 cells increased sharply. The change in A. debilis-induced MMP is shown in Figure 3B, and the MMP decreased gradually. The loss of MMP increased from 11.96% to 66.23% when the cells were treated with 200 μg/mL A. debilis extract. In addition, the expression of SOD2, an ROS balance conditioner, decreased in a dose-dependent manner (Fig. 3C). Relative band intensity was estimated using image analysis software.

ROS content, MMP change, and SOD-2 expression in A. debilis-stimulated HT-29 cells.
Effect of NAC on ROS production and cytotoxicity caused by the A. debilis extract
NAC was used as an ROS scavenger when measuring cytotoxicity and ROS levels to demonstrate that ROS play an important role in A. debilis-induced cell death. Cells treated with the A. debilis extract in the presence and absence of NAC showed different absorbance percentages (Fig. 4A). The A. debilis extract inhibited cell proliferation in a time-dependent manner, and NAC reduced A. debilis-induced cell death. In addition, the amount of ROS increased in the control groups as time passed (Fig. 4B). NAC (2 mM) reduced A. debilis-induced ROS production. Moreover, NAC affected SOD2 gene expression (Fig. 4C). SOD2 expression was higher in the NAC- and A. debilis-treated groups than that in the A. debilis-treated groups. In contrast, SOD2 expression increased in A. debilis cells treated for 6 hours.
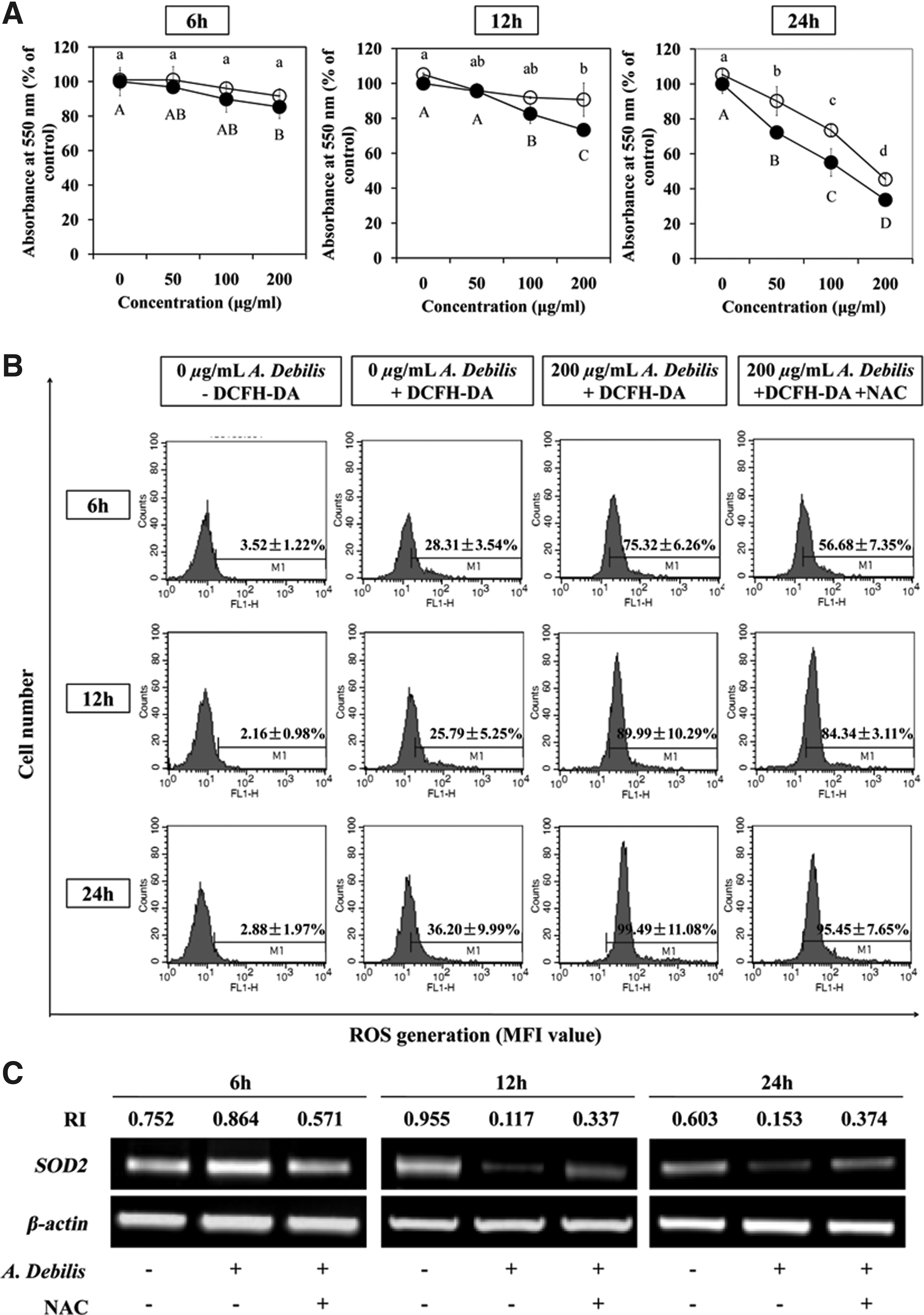
Effect of NAC on ROS production and cytotoxicity caused by A. debili.
Effect of the A. debilis extract on Bax and Bcl-2 expression
Bax and Bcl-2 expression was assessed by RT-PCR and Western blot analyses. As a result, the A. debilis extract upregulated gene Bax expression, but downregulated Bcl-2 expression (Fig. 5). NAC enhanced the A. debilis-induced reduction in Bcl-2 expression (Fig. 6A) and reduced the A. debilis-stimulated Bax/Bcl-2 ratio (Fig. 6B). We also observed increased Bax protein levels in cultures treated with 50, 100, and 200 μg/mL of the extract after 24 hours, concomitant with decreased Bcl-2 expression. The Bax/Bcl-2 ratio increased sharply from 0.83 to 20.83 (Fig. 5D). These results revealed that the clear Bax overexpression and increased Bax/Bcl-2 ratio accelerated apoptosis induced by the A. debilis extract in the HT-29 cell line.
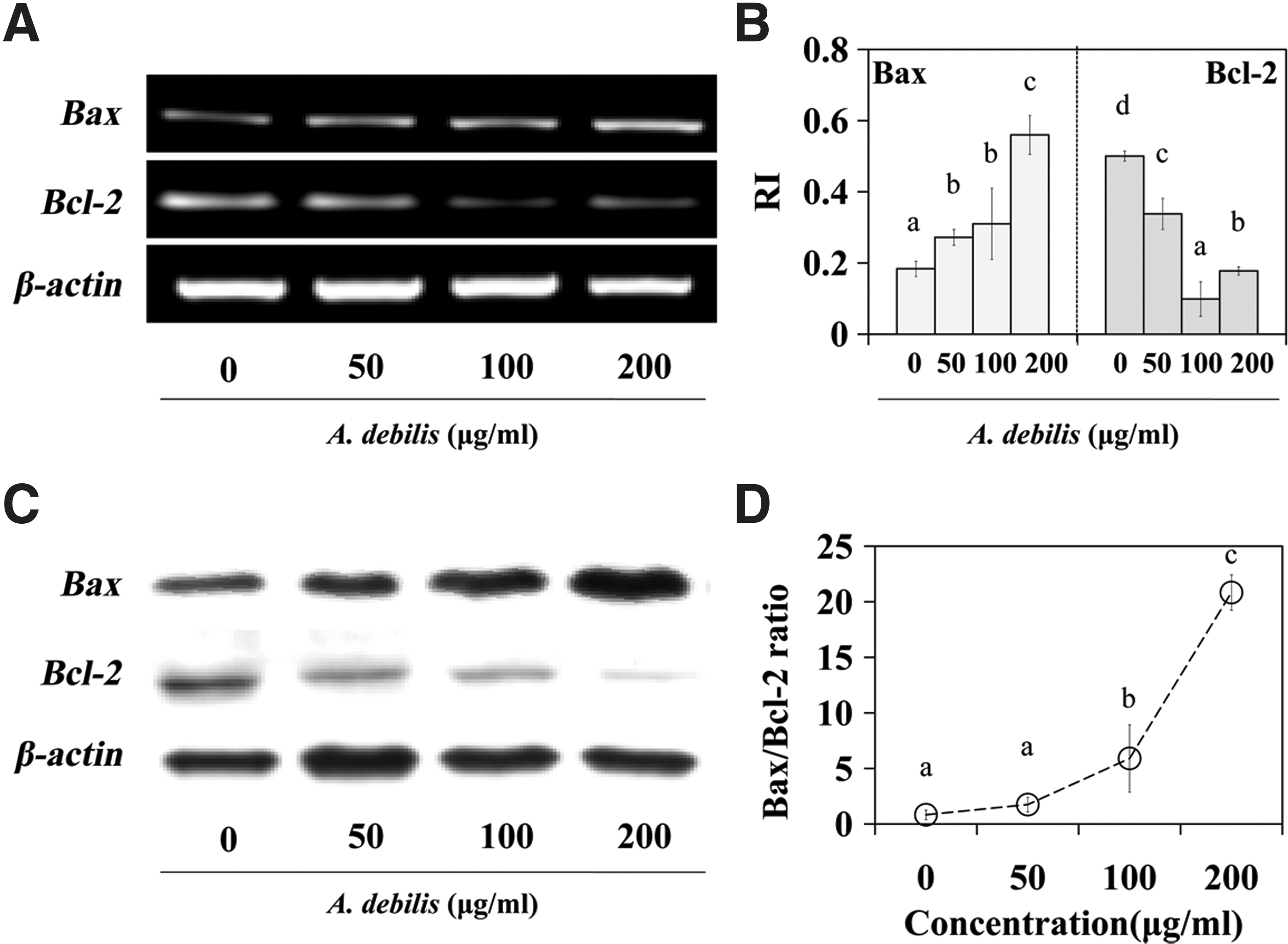
Bax and Bcl-2 expressions in A. debilis-stimulated HT-29 cells.
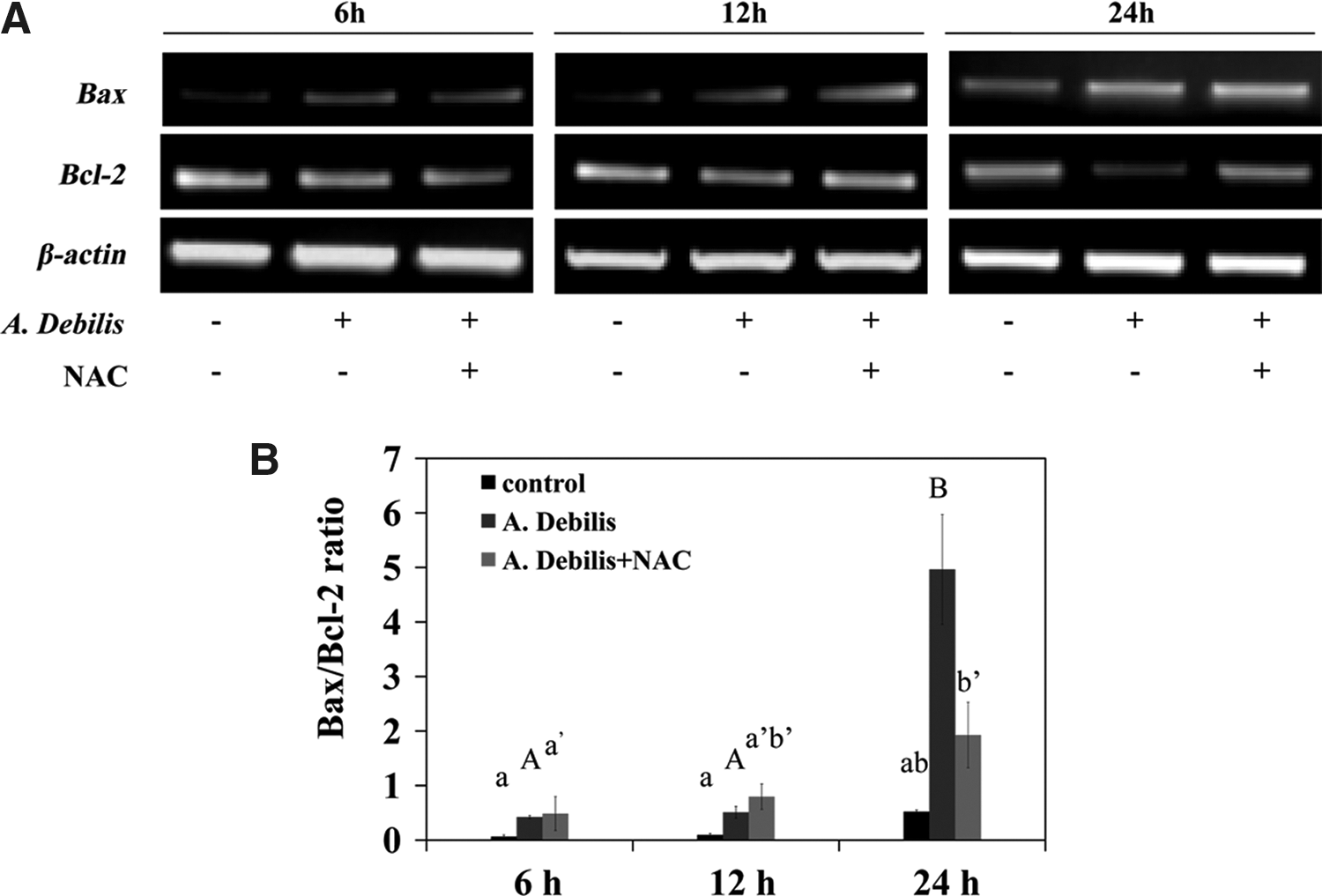
Bax and Bcl-2 expressions in A. debilis-stimulated HT-29 cells with/without NAC.
Discussion
Cancers as a group account for approximately 13% of all deaths worldwide. Normally, cancer can be treated by surgery, radiation therapy, and chemotherapy. 15 Although, many anticancer drugs have been developed, the use of most of them is limited due to their toxicity to normal cells and tissues. 16 Because an overdose of the Aristolochia plant could lead to serious renal failure, 13 we used the normal human embryonic kidney cells (HEK293) as the target to determine the optimal dosages of the A. debilis extract for further research. The results indicated that, dosages ≤200 μg/mL were slightly toxic to HEK293 cells. Similarly, a low dose of A. debilis is not nephrotoxic. 4 Thus, 50, 100, and 200 μg/mL of the A. debilis extract were used in this study. The A. debilis extract inhibited HT-29 cell proliferation, the morphology of the cells changed, and cell numbers decreased.
Because the A. debilis extract decreased HT-29 cell proliferation, we further investigated whether this effect was due to cell cycle arrest. Normally, cells undergo four successive stages such as G1, S (DNA replication), G2, and M (mitosis). G0 is a resting phase in which the cell stops dividing. 17 The A. debilis extract downregulated cells in the G0/G1 phase; however, it upregulated cells in the sub-G1 phase. Most cancer drugs induce cancerous DNA damage, block mitosis, and arrest cells at the G2/M phase. 18,19 However, when apoptotic cells have lost sufficient DNA, the sub-G1 peak will appear in a DNA histogram. 20 Therefore, the apoptosis and cell death induced by the A. debilis extract may have been related to sub-G1 arrest in HT-29 cells.
ROS have been proposed to play an important role in cancer, arthritis, and neurodegenerative disorders. 21 A moderate increase in ROS promotes cell proliferation and differentiation, whereas excessive amounts of ROS cause oxidative damage to lipids, proteins, and DNA. 22 Increased ROS may play an important part in the initiation and progression of cancer through ROS-generating anti-neoplastic therapies. 23 However, excessive levels of ROS and increased oxidative stress can also be toxic to cancer cells. In the present study, ROS increased sharply in A. debilis-treated HT-29 cells. To demonstrate whether ROS was important in A. debilis-induced apoptosis and cell death, the antioxidant NAC was used in control experiments. NAC, a sulfidryl antioxidant, is widely used in basic science research. It protects against oxidative stress-induced cell death, prevents cancer, treats cardiovascular diseases, and chelates heavy metals. 24,25 NAC decreased A. debilis-induced ROS accumulation and increased cell proliferation, which may have been due to decreased ROS and less oxidant stress, resulting in less cell death. Consequently, A. debilis-induced apoptosis and cell death appeared to be associated with the generation of ROS.
Mitochondria produce most ROS by cellular metabolism. Accumulated ROS may cause peroxidative damage of the cell membrane and change membrane permeability. An important indicator of ROS-induced apoptotic mechanism is decreased MMP of injured mitochondria. 26 In contrast, cells balance ROS generation using ROS-scavenging systems such as SODs, glutathione peroxidase, peroxiredoxins, glutaredoxin, thioredoxin, and catalase. SOD2 or MnSOD is found mainly in the mitochondrial matrix. A major intracellular form of the SOD enzyme is considered a tumor suppressor that acts indirectly via ROS. 8 In this study, we investigated SOD2 expression by RT-PCR. In the 6-hour-treated group, the A. debilis extract stimulated SOD2 overexpression. The upregulated SOD2 works as a mechanism to counteract increased ROS stress in the short term. 8 However, as time passed, the increased ROS exceeded SOD2 capacity. The observed decrease in SOD2 expression in the 12- and 24-hour-treated groups may have been associated with the unregulated ROS production in cancer cells. Namely, the downregulation of SOD was due to the increased scavenging. Additionally, ROS inflict direct damage to the cell membrane, change the MMP level and mitochondrial function, and consequently affect DNA expression and enzyme activity. 27 In a vicious circle, the inhibited enzyme activity results in severe accumulation of ROS, resulting in irreversible cellular injury that ultimately results in cancer cell death. 28
The most widely studied genes involved in apoptosis are the Bcl-2 family members. The Bcl-2 family of proteins (proapoptotic protein, Bax and anti-apoptotic protein, Bcl-2), which is located in the mitochondrial outer membrane, plays a role controlling the progress of apoptosis. 29 In this study, we detected Bax and Bcl-2 expression by RT-PCR and Western blot. Bax expression increased, whereas Bcl-2 expression decreased in dose-dependent manner with a sharply increasing Bax/Bcl-2 ratio due to both mRNA and protein levels. These experiments revealed that the clear Bax overexpression accelerated apoptosis induced by the A. debilis extract in HT-29 cells. Ye et al. suggested that the Bax/Bcl-2 ratio is a predictive marker that determines sensitivity to apoptotic stimuli in cancer. 30 An increased Bax/Bcl-2 ratio may play an important role regulating the progression of apoptosis in A. debilis-stimulated HT-29 cells. Li et al. reported that ROS induce apoptosis in cancer cells by suppressing Bcl-2 expression and increasing Bax expression. 31 In our study, differences in Bax and Bcl-2 expression at 6 and 12 hours were unclear. However, NAC significantly downregulated the Bax/Bcl-2 ratio at 24 hours. These results indicate a role for ROS in A. debilis-induced apoptosis of HT-29 cells.
In conclusion, an A. debilis extract showed dose and time effects for inhibiting proliferation and inducing apoptosis in HT-29 cells due to overproduced ROS. Subsequent to the increase in ROS and sub-G1 arrest, A. debilis-incubated cells lost the MMP, showed a decreased SOD2 activity, and an increased Bax/Bcl-2 ratio. In vivo experiments are currently underway to investigate the detailed mechanism of A. debilis-induced apoptosis.
Footnotes
Acknowledgment
This study was carried out with the support of “Indigenous Crops Research & Development Project (Project No. C1009413-01-01)” provided by the Korea Rural Development Administration.
Disclosure Statement
No competing financial interests exist.