
Abstract
Introduction:
Gastrin-releasing peptide receptors (GRPR) and GRP-derived analogs have attracted attention due to high receptor expression in frequently occurring human neoplasia. The authors recently synthesized a series of GRPR-affine peptide analogs based on the 27-mer GRP and derivatized with the DOTA chelator at the N-terminus for 111In-labeling. In this study, the authors evaluated the most promising from these series, DOTA-GRP(13–27), after radiolabeling with 177Lu for future therapeutic applications. In addition, to improve in vivo stability of the peptide against in vivo degradation by the protease neutral endopeptidase (NEP), the authors coinjected [177Lu]DOTA-GRP(13–27) with the potent NEP inhibitor phosphoramidon (PA). The authors also aimed at reducing renal uptake by coadministration of lysine.
Methods:
In vivo stability studies were performed in Swiss albino mice. Biodistribution studies were conducted in NMRI nu/nu mice bearing prostate cancer (PC)-3 xenografts. Ex vivo autoradiography was performed using frozen sections from PC-3 xenografts and kidneys.
Results and Discussion:
Coadministration of PA significantly increased the percentage of intact radiopeptide in the mouse circulation. From biodistribution and ex vivo autoradiography studies, coadministration of both lysine and PA with [177Lu]DOTA-GRP(13–27) appeared to induce a clear improvement of tumor uptake as well as lower levels of renal radioactivity, causing a promising ninefold increase in tumor/kidney ratios.
Introduction
A striking growth in the development of radiolabeled peptide analogs, binding to tumors overexpressing their cognate receptors with high affinity and specificity, has been witnessed during the last two decades. 1,2 Promising preclinical and clinical data obtained for a large number of radiolabeled peptide analogs have paved the way for improved diagnostic imaging through either single-photon emission computed tomography (SPECT) or positron emission tomography (PET) and/or peptide receptor radionuclide therapy (PRRT). The prototypes of such peptides comprise somatostatin analogs targeting their receptors on neuroendocrine tumors (NET), especially somatostatin receptor subtype 2 (sst2). 3,4 Octreoscan®, a 111In-labeled peptide, has been FDA-approved since 1994 for imaging of sst2-positive NET. Second- and third-generation somatostatin analogs combined with the therapeutic radionuclides 90Y (Eβ max 2 MeV, half-life: 64 hours) or 177Lu (Eβ max 0.497 MeV, half-life: 6.7 days) have produced most successful radionuclide therapy agents such as [90Y-DOTA,Tyr3]octreotide (90Y-DOTATOC) and [177Lu-DOTA,Tyr3]octreotate (177Lu-DOTATATE). 5 –7
Another peptide receptor family that received attention is that of the gastrin-releasing peptide receptor (GRPR), a subtype of the bombesin (BBN) receptor family. The presence of GRPR is well documented on a number of different human cancers, such as prostate cancer (PC), breast cancer (BC), and small-cell lung cancer (SCLC). 8 –10 These findings stimulated other groups and us to develop a series of BBN/GRP analogs modified at the N-terminus with a suitable chelate moiety to complex diagnostic and/or therapeutic radionuclides for targeting GRPR-positive tumor sites. 11 –13 Analogs that have been investigated in preclinical and clinical studies on their promise for radionuclide therapy included AMBA [DO3A-CH2COG-4-aminobenzoyl-Q-W-A-V-G-H-L-M-NH2), DOTA-8-AOC-BN(7–14)NH2 (8-AOC=ω-NH2(CH2)7COOH], and DOTA-PESIN (DOTA-15-amino-4,7,10,13-tetraoxapentadecanoic acid-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2) labeled with either 177Lu or 213Bi (Eαmax 5.982 MeV, half-life: 45.6 minutes). 14 –16 Initially, 177Lu-AMBA seemed to be a very promising candidate; it displayed high receptor affinity (Kd=1.02 nM) and a high internalization rate in cells expressing GRPR. Further preclinical studies revealed a most suitable pharmacokinetic profile and a significant inhibition of tumor growth in PC-3 tumor-bearing mice following therapy. In phase I clinical trials though, an undesirably strong uptake in the pancreas was observed and, most importantly, a low plasma stability. 17
In search of novel GRPR-seeking radiotracers, the authors have recently developed a series of highly affine GRP-based receptor agonists truncated in four different positions and modified with the 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelator for stable binding of diagnostic (111In) or therapeutic (177Lu) radionuclides. Within this series [111In]DOTA-GRP(13–27), a Lys 13 -containing analog had a high GRPR affinity (IC50=2.3 nM) and the highest internalization rate in GRPR-positive cells. Furthermore, it showed the highest uptake in PC-3 xenografts in SCID mice. However, renal uptake and retention values were very high (≈46%ID/g at 4 hours postinjection [p.i.] and ≈30%ID/g at 24 hours p.i.), an unfavorable feature during radionuclide therapy due to the risk of long-term renal damage. Moreover, in vivo stability studies in Swiss albino mice revealed rapid degradation of the radiopeptide (≈20% intact peptide at 5 minutes p.i.). 18 This radiotracer, once exposed to proteolytic enzymes within the body, was rapidly degraded due to the higher number of possible cleavage sites in the natural peptide motif compared to the GRP(18–27) peptide analog ([99mTc]Demomedin C). 19
Increased resistance to such proteolytic activity might translate into prolonged circulation of radiolabeled peptide analogs preferably leading to higher receptor targeting as well as higher tumor uptake of radioactivity in tumor lesions. One of the most prominent proteases in the degradation process of radiopeptides from different peptide families is the enzyme neutral endopeptidase (NEP, EC 3.4.24.11). 20 –22 Phosphoramidon (PA), a natural product first isolated from cultures of Streptomyces tanashiensis, is a potent competitive inhibitor of NEP. Its water-solubility, potency, commercial availability, and low costs, render PA an excellent candidate for these purposes. 23 –26 The authors were interested in exploring the effects of PA against in vivo degradation of radiopeptide analogs originating from a variety of peptide families. This innovative method showed promise regarding improved in vivo stability of peptide analogs from different peptide families, including a GRP analog targeting GRPR in pilot studies, as recently published by these groups. 27 With the aim to translate these findings to other peptide analogs, the authors applied the novel approach of PA coadministration to enhance proteolytic DOTA-GRP(13–27) resistance by enzyme inhibition.
The undesirably high renal uptake of 111In-labeled DOTA-GRP(13–27) represents an obstacle for therapeutic applications. Several peptide derivatives have been published to be reabsorbed after glomerular filtration into the renal proximal tubules. 28 For a number of peptide radiotracers, it has been documented that megalin, a 600-kDa transmembrane multiligand low density lipoprotein (LDL)-protein, plays a key role in the retention of such radiopeptides within the kidney. 29 –32 Different strategies can be applied to reduce the renal radiation dose in patients by interfering with the megalin system. 33 –36 The techniques most widely used is that of basic amino acid coadministration to reduce renal reabsorption of the radiopeptide. Specifically, it has been reported for radiolabeled somatostatin analog therapy that coadministration of the positively charged amino acids, lysine (Lys) and arginine (Arg), effectively competes with the tubular reabsorption of radiolabeled somatostatin analogs containing positively charged amino acids. 37,38 Since [177Lu]DOTA-GRP(13–27) contains two basic amino acid residues, Lys 13 and Arg 19 , coinjection of the Lys solution was expected to reduce the renal accumulation of this radiolabeled compound as well. In the current study, the GRP analog [177Lu]DOTA-GRP(13–27) was preclinically evaluated for future application in targeted radionuclide therapy of GRPR-expressing human tumors in receptor-positive, tumor-bearing mice. Aiming at a biodistribution with a favorable tumor-to-kidney ratio, the authors implemented the innovative strategy of coinjecting the NEP inhibitor (PA) as well as Lys with the radiotracer to increase GRPR targeting in combination with renal uptake inhibition.
Materials and Methods
Reagents
Unless otherwise stated, chemicals were reagent grade and used without further purification. The analog of mammalian GRP [DOTA-GRP(13–27)=DOTA-Lys-Met-Tyr-Pro-Arg-Gly-Asn-His-Trp-Ala-Val-Gly-His-Leu-Met-NH2], was purchased from Pichem (Graz, Austria). 177LuCl3 was purchased from IDB (Baarle Nassau, The Netherlands). Quenchers added included ascorbate (Bufa BV, Uitgeest, The Netherlands), gentisic acid (Covidien, Petten, The Netherlands), and
177Lu-labeling
The lyophilized peptide conjugate was dissolved in high-performance liquid chromatography (HPLC)-grade water at a final 1 mM concentration. Labeling was conducted in double-sealed polypropylene reaction tubes (PCR thermocycler tubes, maximal volume 150 mL, MoBiTec; ITK Diagnostics, Uithoorn, The Netherlands), by addition of 150 MBq 177LuCl3 (IDB) to 7.5 nmol peptide (20 MBq/nmol) and incubation of the mixture in a sodium acetate buffer, pH 4.5 (Vf=140 μL), for 15 minutes at 80°C. After labeling, quenchers were added to prevent oxidation and radiolysis (4 mM ascorbic acid, 4 mM gentisic acid, and 50 mM
Cell culture
The human androgen-independent prostate cancer cell line PC-3 was cultured at 37°C in a humidified atmosphere containing 5% CO2 in the RPMI medium (Lonza, Vierviers, Belgium) supplemented with 5% fetal calf serum (Gibco Invitrogen Co., Grand Island, NY) and 5 mL/500 mL penicillin/streptomycin antibiotics (10,000 U/mL penicillin, 10,000 U/mL streptomycin; Lonza). Cells were cultured until near confluence and a trypsin/EDTA (170,000 U/L Trypsin-Versene and 200 mg/L EDTA) solution (Lonza) was used for weekly 1:3 or 1:4 passages.
Internalization of [177Lu]DOTA-GRP(13–27) in PC-3 cells
PC-3 cells were seeded in six-well plates (≈1–1.5×106 cells/well) before the day of the experiment and reached confluence after 24 hours of incubation. Internalization studies were performed, as described previously, 19 by using ≈40,000 cpm of 177Lu-radioconjugate corresponding to 250 fmol total peptide in 150 μL of 0.5% bovine serum albumin/phosphate-buffered saline (PBS). Incubation in the presence of 1 μM final concentration of [Tyr4]BBN was performed to determine nonspecific internalization. Medium sample radioactivity was measured in a gamma counter (LKB-1282-compugamma system; PerkinElmer, Groningen, The Netherlands) and results were calculated as percent internalized per total added activity per million cells and represent the average of at least two experiments performed in triplicate.
In vivo stability of [177Lu]DOTA-GRP(13–27)
For metabolic evaluation assays, 40–60 MBq of 177LuCl3 was added to 3 nmol total peptide in the labeling solution (specific activity [SA]: 13–20 MBq/nmol), and the test radiotracer was injected as bolus (100 μL, in saline, vehicle) together with vehicle (100 μL, control A) or PA (100 μL of vehicle containing 300 μg PA, PA treated B) in the tail vein of male Swiss albino mice (30±5 g, from NCSR Demokritos Animal House Facility). After 5 minutes, blood was collected from the heart of the animals under anesthesia and immediately transferred into prechilled polypropylene tubes, containing EDTA, and placed on ice. Blood samples were centrifuged (10 minutes, 2000 g/4°C; in a Hettich, Universal 320R, centrifuge, Tuttlingen, Germany), plasma was collected, mixed with chilled ACN in a 1:1 v/v ratio, and centrifuged again (10 minutes, 15,000 g/4°C). Supernatants were concentrated to a small volume under a gentle N2-flux at 40°C, diluted with saline (≈400 μL), and filtered through a Millex GV filter (0.22 μm). Aliquots were analyzed by HPLC (system B). The elution time of intact [177Lu]DOTA-GRP(13–27) was determined by coinjection with a sample of the labeling reaction solution.
Biodistribution in PC-3 tumor-bearing mice
Approximately 6×106 freshly harvested human PC-3 cells in 150 μL PBS were subcutaneously inoculated in the right shoulder of 6- to 7-week-old NMRI nude mice (Harlan, Horst, The Netherlands). The animals were kept under aseptic conditions and well-palpable tumors developed within 3–4 weeks at the inoculation site. The effect of peptide dose escalation [in a range of 0, 250, 500, and 1000 pmol total unlabeled peptide mass coinjected with 10 pmol of [177Lu]DOTA-GRP(13–27)/animal] on the uptake in the organs of interest, such as the pancreas and tumor, was investigated. On the day of the experiment, 4 groups (4 animals/group) received different peptide amounts as a 100 μL bolus with [177Lu]DOTA-GRP(13–27) (specific activity: 20 MBq/nmol) through the tail vain (10 pmol/mice), and animals were sacrificed at 24 hours p.i. To investigate the effects of PA and Lys, biodistribution studies were conducted with each animal receiving 100 μL bolus of radiopeptide (0.2 MBq/10 pmol) together with either vehicle (100 μL, control; group A), Lys (100 μL of vehicle containing 20 mg Lys; group B), PA (100 μL of vehicle containing 300 μg PA; group C), or a combination of the two (100 μL of vehicle with Lys+PA; group D). After 24 hours, the organs of interest were collected, weighed, and measured for their radioactivity content using a shielded well-type gamma counter (Wizard, Wallac). Biodistribution data were calculated as percentage of injected dose per gram (%ID/g) tissue using standards of the injected dose. The kidneys and tumors were processed further for ex vivo autoradiography (vide infra).
All animal experiments were carried out in compliance with European and national regulations and after approval of protocols by local authorities.
Ex vivo autoradiography
After isolation of the PC-3 tumors and kidneys, the Erasmus MC group embedded the tissues in the Tissue-Tek® optimum cutting temperature (O.C.T.) compound (Sakura Finetek, AJ Alphen aan den Rijn, The Netherlands) and frozen at −80°C. Sections of 10 μm were cut using a cryostat (Cryo-Star HM 560 M; Microm, Walldorf, Germany), mounted on slides (Menzel, Braunschweig, Germany), and exposed for 24–72 hours to SR phosphor imaging screens (Packard Instruments Co., Meriden, CT) in X-ray cassettes. Screens were read by a Cyclone Phosphor Imager and analyzed with the OptiQuant 03.00 image processing system (PerkinElmer).
Statistics
Data from biodistribution studies are expressed as mean±standard deviation, statistical analysis was performed using the Kruskal–Wallis test, and multivariate ANOVA with the Bonferroni post hoc test. These tests were confirmed with Dunnett's two-sided test.
Results
177Lu-labeling
[177Lu]DOTA-GRP(13–27) efficiently formed by heating DOTA-GRP(13–27) together with 177LuCl3 in an acidic medium. The radiotracer was produced in high specific activities (20 MBq/nmol) with a radiochemical yield of ∼93% as verified by ITLC and reverse-phase HPLC (RP-HPLC) (Fig. 1; system A, column I). The radioligand was used without any further purification step.

Radiochromatogram of reverse-phase high-performance liquid chromatography (RP-HPLC) analysis of [177Lu]DOTA-GRP(13–27)-labeling reaction mixture (system A, column I).
Internalization of [177Lu]DOTA-GRP(13–27) in PC-3 cells
The internalization rate of [177Lu]DOTA-GRP(13–27) in PC-3 cells after a 1-hour incubation at 37°C was high (≈27% of total added activity was internalized, only 6% was bound to the cell membrane). The percentage of the internalized radiotracer declined to base levels in the presence of 1 μM [Tyr4]BBN, indicating a specific GRPR-mediated process.
In vivo stability of [177Lu]DOTA-GRP(13–27) in vivo
RP-HPLC analysis of mouse blood samples, collected 5 minutes after a bolus injection of the compound in vivo, revealed the presence of only ≈30% intact peptide in the blood. However, PA coinjection resulted in a substantial increase in circulating intact peptide to 63%, as illustrated in Figure 2 (system B, column II).
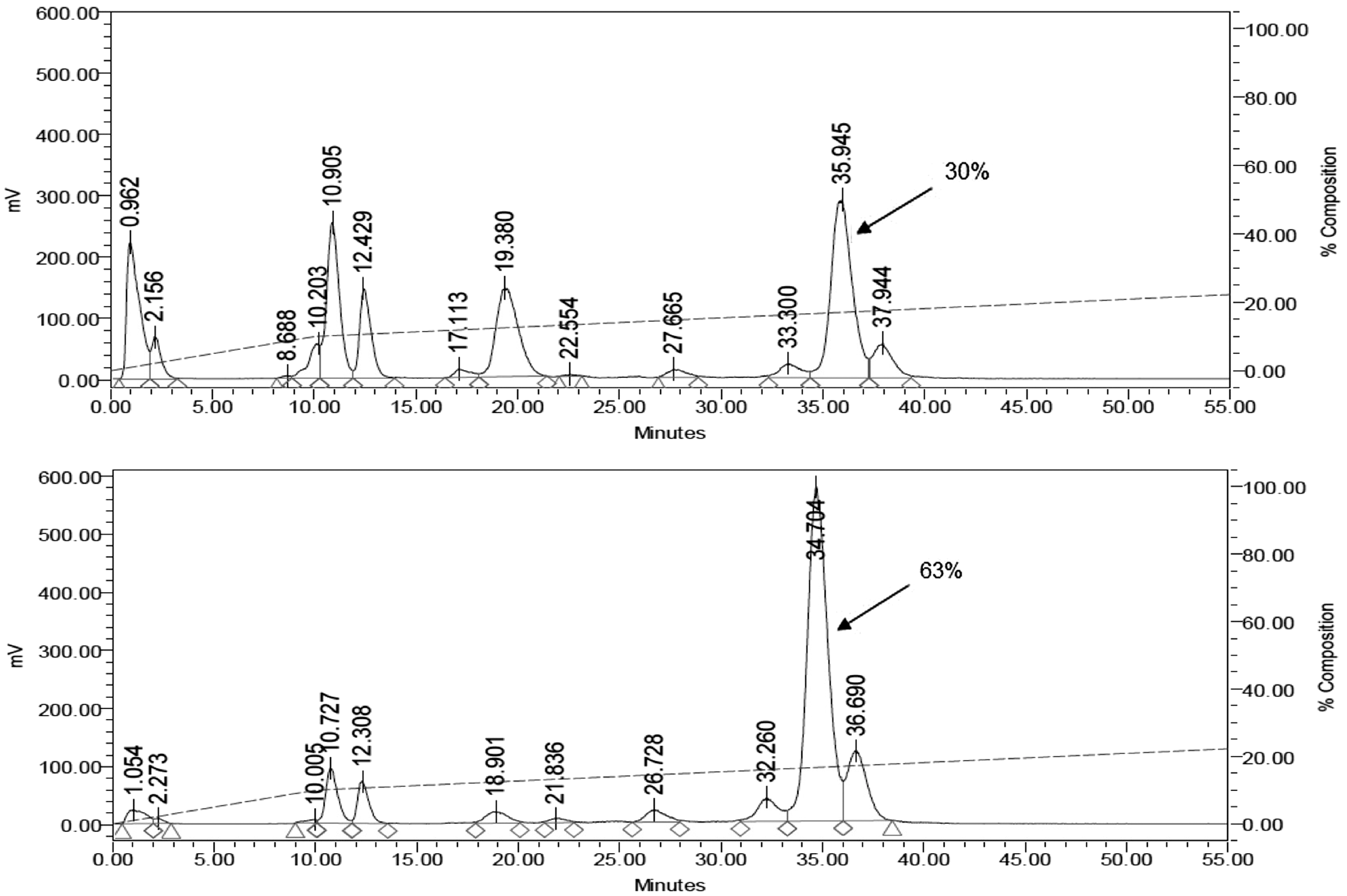
RP-HPLC analysis radiochromatogram of mouse blood collected 5 minutes postinjection (p.i.) of
Biodistribution in PC-3 tumor-bearing mice
An escalation study using different doses from 0 up to 1000 pmol of unlabeled peptide was performed by coinjection with 10 pmol [177Lu]DOTA-GRP(13–27) in NMRI nu/nu mice bearing PC-3 human prostate cancer xenografts. In GRPR-expressing tissues such as the pancreas, a reduction of uptake was observed with increasing peptide doses. More specifically, pancreatic uptake displayed a decline up to 4.5% at the highest peptide amount, resulting from partial saturation of available binding sites. Interestingly, no significant differences were observed in tumor uptake while increasing the administered unlabeled peptide dose. Radioactivity was cleared from the body almost exclusively through the kidneys into the urine (Fig. 3). In accordance with the in vivo blood stability results, animals that received PA coinjection showed a striking increase in uptake in both receptor-positive xenografts and pancreas (Tumor: from 1.3%ID/g to 4.0%ID/g; Pancreas: from 17.4%ID/g to 50.8%ID/g). Lys treatment did not significantly influence the uptake of the radiopeptide in PC-3 tumor and the GRPR-expressing pancreas compared to control, while renal uptake was reduced by ≥50% (from 39.3%ID/g to 16.5%ID/g). Coadministration of both Lys and PA (group D) resulted in an improvement in the tumor-to-kidney ratio by a factor of 9 (from 0.03 to 0.28). Tissue distribution data and kidney-to-tumor ratios at 24 hours after administration are summarized in Table 1 and Figure 4.
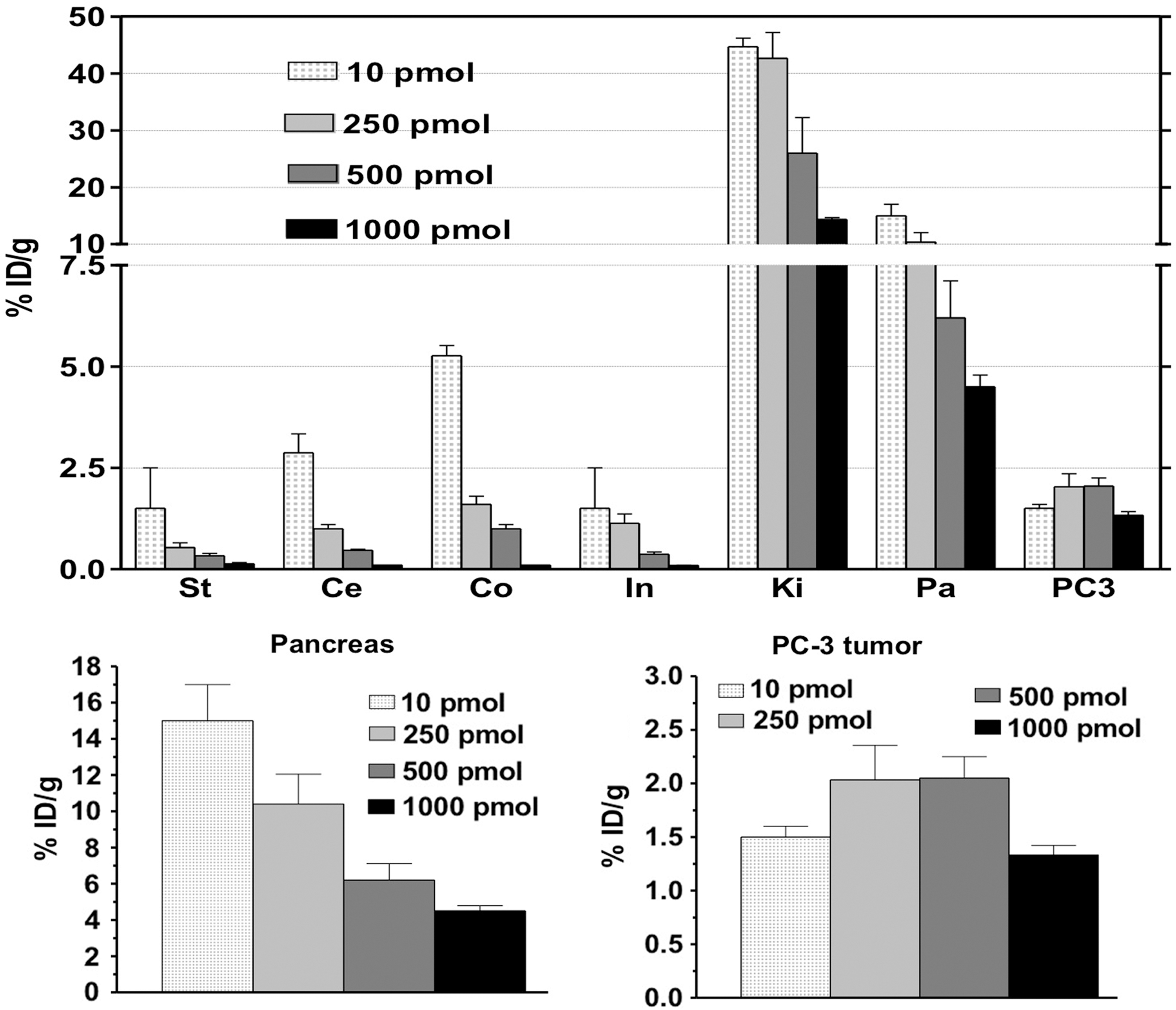
Tissue distribution data of 10 pmol [177Lu]DOTA-GRP(13–27), supplemented with 0, 50, 500, and 1000 pmol GRP(13–27) in prostate cancer (PC)-3 tumor-bearing NMRI nu/nu mice at 24 hours p.i. for St, stomach; Ce, cecum; Co, colon; In, intestines; Ki, kidneys; Pa, pancreas; and PC-3, PC-3 xenograft.

Tissue distribution data of 10 pmol [177Lu]DOTA-GRP(13–27) at 24 hours p.i. in PC-3-tumor-bearing NMRI nu/nu mice for (I); tumor (II); pancreas, and (III); kidneys; ***P<0.001.
Biodistribution uptake values are given as mean±standard deviation in %ID/g.
Lys, 20 mg of lysine in 100 μL bolus of [177Lu]DOTA-GRP(13–27); Pa, 300 mg of phosphoramidon in100 μL bolus of [177Lu]DOTA-GRP(13–27); Lys+PA, the aforementioned quantities in 100 μL bolus of [177Lu]DOTA-GRP(13–27); PC, prostate cancer.
Ex vivo autoradiography
Representative autoradiograms of renal sections as presented in Figure 5A showed a clear reduction in renal cortical radioactivity in the mice treated with Lys, as compared to the untreated controls, whereas PA+Lys cotreated animals displayed similarly low radio-accumulation in the kidneys as those in the Lys group. The localization of retained radioactivity in the PC-3 tumor, as shown in Figure 5B, appeared to be homogenous, with highest intensity in the Lys+PA group in line with the biodistribution results.

Ex vivo autoradiograms of 10 μm slices of mouse kidney
Discussion
Following the paradigm of radiolabeled somatostatin analogs for tumor scintigraphy and radionuclide therapy, a rapid development of GRPR-avid radiolabeled peptide ligands used for imaging and therapy on frequently occurring types of cancer such as PC, SCLC, and BC has been witnessed during the last years. 8 –10 Stability of radiotracers in the circulation is of crucial importance for delivery at tumor lesions. In an attempt to prevent rapid degradation of radiopeptides by peptidases within the body, widely used approaches include the structural modification of such peptide analogs. In previous studies, this group among others has shown the effects of substitution of native chain residues in the GRP(18–27) sequence with, for example, unnatural amino acids and the effects of such changes on receptor affinity, in vivo stability, and tumor uptake of corresponding [99mTc]-radiotracers. 19,41 –43 The main and most obvious disadvantages of this approach appeared to be a potentially decreased affinity for the different receptor subtypes and, in general, a compromised pharmacokinetic profile of the analog. As an alternative to the usually followed methods of peptide-chain derivatization, the authors pursued a novel approach of in vivo enzyme inhibition with [177Lu]DOTA-GRP(13–27). This peptide analog was proven to be among the best from a series of DOTA-chelated truncated analogs of the human 27-mer GRP, as recently published. 18 However, the low in vivo stability was an important obstacle to overcome. The ubiquitous enzyme NEP 24.11, a zinc-dependent membrane-bound metalloprotease responsible for the degradation of many native peptides, including bombesin and GRP, seemed to be a suitable candidate for in vivo intervention. 21,22 PA is a potent, competitive, and reversible inhibitor of NEP, exhibiting an in vitro IC50 value of 10 nM. 23,24 Coinjection of PA has been recently shown to increase the in vivo stability of radiolabeled peptide analogs from the somatostatin, bombesin, and gastrin family. As a result, PA had a profound influence on radiotracer uptake in receptor-positive xenografts in vivo. 27 Motivated by these results, the authors aimed on reducing the in vivo degradation of [177Lu]DOTA-GRP(13–27) as well. To examine the in vivo stability of [177Lu]DOTA-GRP(13–27), the authors applied an optimized in vivo blood assay, which is preferable over the usually applied in vitro incubations in serum. The latter often leads to overestimation of peptide stability as it does not take into account the contribution of NEP anchored to blood vessels and in major organs in the body, as well as in the tumor-surrounding matrix. 44 For this purpose, the authors monitored the levels of intact circulating [177Lu]DOTA-GRP(13–27) as well as the changes induced by PA treatment. The significant increase of intact peptide in the blood observed after PA coadministration suggests a prominent role for NEP in the degradation of [177Lu]DOTA-GRP(13–27). Yet, more than one enzyme may be involved in the degradation process, a hypothesis that aligns with the results of metabolic studies reported for [177Lu]AMBA. 17 In agreement with these mouse in vivo blood stability data, PA coinjection resulted in an equally favorable improvement on the pharmacokinetic profile during biodistribution studies in mice. Biodistribution results of [177Lu]DOTA-GRP(13–27) matched the results obtained with the 111In-labeled counterpart. 18 After coadministration of PA, a high GRPR-specific uptake in the tumor was observed, while the background activity remained low, even at 24 hours p.i.; an attractive property for PRRT. On the other hand, the observed high renal uptake values hindered further translational investigations. As the DOTA-GRP(13–27) sequence contains two positively charged residues, the authors coinjected the Lys solution in PC-3-bearing mice in an attempt to reduce the renal accumulation to this dose-limiting organ by competition, as also performed in the clinic for reduction of renal uptake of somatostatin analogs. 38 Lys coadministration resulted in more than 50% decrease of renal reabsorption of the radiotracer without significant effect on tumoral or pancreatic uptake. Finally, coadministration of both agents, PA and Lys, together led to a most promising effect on human xenograft uptake that significantly increased, while renal reabsorption was significantly reduced to levels comparable to those obtained after Lys coinjection alone. The biodistribution data of the (Lys+PA) animal group show that the effect of Lys on renal uptake and that of PA on pancreas and tumor uptake remained practically unaffected when these two agents were coadministered. The i.v. administration route in mice in quantities of up to 300 μg PA showed no adverse effects caused by this inhibitor. Moreover, PA elicited its protective properties after a single coadministration, which is considered an important asset for future clinical use. The authors expect this innovative strategy of enzyme inhibition in vivo, combined if needed with renal protection agents, to be of considerable benefit for molecular imaging and targeted therapy with a number of biodegradable radiopeptides. Based on the aforementioned results, it can be predicted that this, in turn, will lead to increased diagnostic sensitivity and higher therapeutic efficacy. More specifically, in the field of radionuclide therapy, as PRRT protocols usually consist of a confined number of administrations, potential protection of the radiolabeled analog from degradation until it reaches its designated target is considered to be the most promising to enhance tumor localization.
Next to the effects of PA and Lys, the peptide amount escalation studies revealed a significant decrease in pancreas uptake with increased peptide amounts, while that in human xenografts remains practically unaffected. Indeed, one would expect affected tumor uptake with peptide amount escalation. However, the authors have experienced also in earlier studies that tumor uptake is less influenced by higher peptide amounts than pancreatic uptake and that only at high peptide amounts, tumor uptake decreased. 45 Also, in the current study, with the highest peptide amount some decrease in tumor uptake was observed, resulting in a bell-shaped-like curve, like in other radiotracer studies. 17,46,47
In PRRT, where higher peptide amounts have to be administered to achieve the desired therapeutic effect from the radionuclide attached, this will be an advantage as decreased uptake in the GRPR-positive pancreas in combination with an unaffected retention of radioactivity in tumor lesions would lead to even further improved tumor-to-background ratios.
Conclusions
The innovative coadministration of the NEP inhibitor PA notably enhanced the in vivo stability of [177Lu]DOTA-GRP(13–27) in mice, which translated into a significant increase of in vivo GRPR targeting and tumor uptake. Furthermore, Lys coadministration resulted in an ∼60% reduction of renal uptake and retention. Finally, the combination of both agents revealed a ninefold increase of the tumor-to-kidney ratio, encouraging further research on this exciting approach. The strategy of enzyme inhibition may be applied in a considerable number of peptidic radiotracers, which so far were inefficient due to their low stability in circulation.
Footnotes
Disclosure Statement
No financial conflict of interests exist.