
Abstract
Peptide receptor radionuclide therapy (PRRT) is a site-directed targeted therapeutic strategy that specifically uses radiolabeled peptides as biological targeting vectors designed to deliver cytotoxic levels of radiation dose to cancer cells, which overexpress specific receptors. Interest in PRRT has steadily grown because of the advantages of targeting cellular receptors in vivo with high sensitivity as well as specificity and treatment at the molecular level. Recent advances in molecular biology have not only stimulated advances in PRRT in a sustainable manner but have also pushed the field significantly forward to several unexplored possibilities. Recent decades have witnessed unprecedented endeavors for developing radiolabeled receptor-binding somatostatin analogs for the treatment of neuroendocrine tumors, which have played an important role in the evolution of PRRT and paved the way for the development of other receptor-targeting peptides. Several peptides targeting a variety of receptors have been identified, demonstrating their potential to catalyze breakthroughs in PRRT. In this review, the authors discuss several of these peptides and their analogs with regard to their applications and potential in radionuclide therapy. The advancement in the availability of combinatorial peptide libraries for peptide designing and screening provides the capability of regulating immunogenicity and chemical manipulability. Moreover, the availability of a wide range of bifunctional chelating agents opens up the scope of convenient radiolabeling. For these reasons, it would be possible to envision a future where the scope of PRRT can be tailored for patient-specific application. While PRRT lies at the interface between many disciplines, this technology is inextricably linked to the availability of the therapeutic radionuclides of required quality and activity levels and hence their production is also reviewed.
Introduction
Radionuclide therapy is a treatment modality that uses unsealed radioactive sources in the form of a therapeutic radionuclide attached to a targeting vector to deliver therapeutic doses of ionizing radiation to specific disease sites either for curative intent or for disease control and palliation. 1,2 This approach using systemic administration combines the advantage of brachytherapy or external beam radiotherapy and simultaneously behaves like chemotherapy to treat disseminated diseases. 3,4 A systemic administration of radiopharmaceuticals with the known cytotoxic effects of ionizing radiation results in targeting the disease that has spread throughout the body, including malignant cell populations. The success of radionuclide therapy depends on the specificity of the therapeutic targeting agent by virtue of its interaction with a molecular species that is present or absent in a disease tissue to deliver the radiation either to ablate or damage through energetic emissions, such as α-particles, β− particles, or Auger electrons, and conversion electrons. 4 –6 The targeting feature of radionuclide therapy is mainly achieved either by the intrinsic targeting properties of some radionuclides and/or, more commonly, by conjugating the radionuclide to a specific carrier/targeting molecule or vector. The choice of targeting vector is crucial in the development of an effective radiopharmaceutical since the effectiveness of targeted therapies depends on expression of the molecular target.
In light of the need to deliver a cytotoxic radiation dose to the disease site, for example, in the case of tumor, a variety of parameters characterizing the tumor topology, morphology, and physiology need to be considered for selecting the targeting ligand, bifunctional chelating agent (BFCA), and the type of therapeutic radionuclide. While the intrinsic chemical properties of therapeutic radionuclides such as radioiodide (i.e., 131I) can be exploited to target the disease site, examples of physiological accumulation of radionuclides per se in tumors are limited. Tumor-seeking carrier molecules, such as monoclonal antibodies, antibody fragments, peptides, small molecules, liposomes, dextrans, and microspheres, all have different targeting mechanisms and are currently being used or can potentially be used for development of strategies for radionuclide therapy. The essential properties of carrier molecules include biospecificity, in vivo stability, and carrier–target affinity, distribution in target and nontarget sites, excretion, and interaction with the target tissue microenvironment.
In this context, the receptor–ligand paradigm based on the binding of a carrier molecule (the ligand) to a specific site on a target molecule (the receptor) with high affinity is an attractive therapeutic modality. While the presence of multiple binding sites provides the scope for binding more than one ligand, the maximum stoichiometry will always be a low multiple of the number of receptor molecules available. The key determinants of success for this type of therapy rest on the specificity and affinity of the receptor–ligand interaction and the receptor density on the target. This approach constitutes the molecular basis for targeting peptide receptors, which are often expressed in large quantities in certain tumors by small peptides, and has become a very strong focus of interest in applications of such radionuclide therapy.
Over the past decades, there has been tremendous progress in the development of a number of delivery agents and concepts to establish an efficient and reliable method of receptor targeting. Among these, the use of peptides and peptide receptors is of great significance because specific peptide receptors are often expressed on many primary human cancer cells. The high overexpression of these receptors on various tumor cells compared with their low density in normal tissues and extremely high affinities (nano- or subnanomolar range) form the molecular basis for their clinical use as tumor receptor therapeutic agents. Because of their small size, peptides usually exhibit rapid pharmacokinetics and good tumor-targeting characteristics with the ability to efficiently penetrate into tumors.
The importance of peptide receptor radionuclide therapy (PRRT) has generated a number of review articles describing many innovative and exciting developments. 7 –13 The goal of this article is to provide an update on the current key advances in the development of radiolabeled molecular vectors for PRRT from the perspective of basic science, which will serve as a resource for scientists and clinicians involved in the development, evaluation, and application of this therapeutic strategy. This multidisciplinary review is focused on the principles of PRRT, selection criteria of radionuclide and molecular vectors, recent developments and current status, and the key challenges and opportunities for further development.
Regulatory Peptides
Peptides are molecules consisting of two or more amino acids linked together with peptide bonds and usually contain less than 50 amino acids, having a maximum molecular mass of ∼5500 Da. Regulatory peptides are peptide molecules, which regulate or control several biological functions in living organisms. These are either naturally produced by various cells or artificially synthesized, maintaining the same sequence of amino acids, which is responsible for the specific biological function. While these peptides are synthesized as large precursor molecules (for inactive storage), they are subsequently cleaved to smaller active forms or metabolites by specific enzymes or modified by addition of chemical groups. Peptides that are primarily synthesized in the brain, especially in neurons, are called neuropeptides and function either directly or indirectly to modulate synaptic activity. In addition, neuropeptides may also function as primary neurotransmitters. Peptides are also found in the gut, lymphatic tissue, endocrine system, and other organs.
The heterogeneous hormones secreted from cells that line the lumen of the gut and produced by the gastrointestinal tract of mammals not only control digestion but also regulate cell growth. While the function of some peptides are uncertain, such as substance P and pancreatic polypeptide secreted by endocrine tumor cells recognized as gut hormones, they are responsible for some of their systemic manifestations.
Regulatory peptides play an important role in the pathogenesis of several common diseases of the gastrointestinal, respiratory, and urinary tracts, in inflammatory diseases, and even in diseases of the circulatory system. They are known to act on multiple targets in the human body at extremely low concentrations. 14 Targets of these peptides consist of the brain, the gastrointestinal tract, the endocrine system, the kidneys, the lungs, and the immune, vascular, and peripheral nervous systems. They not only regulate and modulate most physiological and metabolic processes but also the function of almost all key organs. Some of the important regulatory peptides that have been used or have the potential for use in PRRT along with their biological effects are depicted in Table 1. 15
Biologically active regulatory peptides are coded by important genes and their targets are proteins or protein-coupled receptors. The action of regulatory peptides is mediated through specific membrane-bound receptors, almost all of which belong to various types of G protein-coupled receptors, and a particular set of downstream target proteins in the cellular plasma membrane.
The peptide receptor-binding event can activate or inhibit biological processes through different mechanisms, including activation of G proteins, tyrosine kinases, or transcription processes. Important for PRRT applications, many of these receptors have been shown to be significantly overexpressed in certain diseases, especially in particular tumor types. The tremendous therapeutic prospect associated with peptides and peptide receptors is the possibility of receptor targeting because peptide receptors are often overexpressed in many primary human cancers. A considerable amount of fascinating research and innovative strategies have thus been focused on understanding the biology of these processes and their application to PRRT. In several instances, it has been demonstrated that peptide receptors are overexpressed in cancer, in comparison with their expression in normal tissue adjacent to the neoplasm and/or in normal tissue of origin. The scope of using these receptors as binding sites for peptide analogs has generated tremendous interest in the development of specific peptides to target these tumors for therapy.
Peptides as Therapeutic Vectors
The ability of regulatory peptides or their analogs to target specific receptors expressed on the surfaces of human cancer cells offers the scope of their use as transport vehicles to guide agents labeled with radionuclides to tissues expressing particular receptors.
10,11
These receptors constitute useful molecular targets for the treatment of cancer owing to their location on the plasma membrane. It is pertinent to point out that this targeting strategy relies primarily on the presence of receptors on tumor cells, which are able to bind with high affinity to the peptide analogs, which is quantified by the respective binding affinity (IC50) values. The desired IC50 value of the radiolabeled peptide analog should be in the nanomolar range. The attractiveness of regulatory peptides as therapeutic vectors in PRRT is primarily based on the following considerations: • The peptides have high affinity and specificity toward a wide range of receptors overexpressed by cancer cells. • The small size and low molecular weight of peptides, compared with proteins and antibodies, facilitate rapid penetration to target tissue. Due to their low molecular weight they show rapid diffusion in target tissue and upon binding of the radioligand, the receptor–ligand complex is generally internalized postbinding, allowing long retention in tumor cells and permitting the emission of ionizing radiation from decay of the attached radionuclide to selectively destroy the targeted cell. • Peptides usually display favorable pharmacokinetics characterized by high concentration in the target tissue and rapid clearance from the blood and nontarget tissue. Peptides are usually rapidly excreted from the body through renal or hepatobiliary excretion or both, depending on the peptide and the type of structural modifications. In fact, the elimination processes can be modified by chemical manipulation to accommodate a variety of possible routes of excretion or metabolism. • Regulatory peptides and their analogs lack antigenicity owing to their small size and hence are nonimmunogenic. • Regulatory peptides are generally nontoxic. • Regulatory peptides exhibit minimal side-effects during therapy because they usually play a modulatory role in various biological systems.
Table 2 summarizes a number of important regulatory peptides and their analogs, which are of current interest for clinical PRRT application targeted to receptors overexpressed on tumors. 7,11 As most of the peptides act through multiple peptide receptor subtypes, it is crucial that the peptide receptor subtype expressed by a given tumor corresponds to the subtype to which the radioligand used for PRRT binds with high affinity.
GRP, gastrin-releasing peptide; NET, neuroendocrine tumor; RGD, arginine-glycine-aspertic acid.
Agonist versus antagonist
The peptide analogs used as the targeting vector to deliver the radiation dose to the diseased site may function both as the receptor agonist or antagonist. An important feature of such agonists is that after binding to the receptor, the peptides or their radiolabeled analogs are internalized by receptor-mediated endocytosis, leading to an accumulation of the radioactive agonist within the tumor cell. 16,17 On the other hand, peptide analogs, which function as receptor antagonists, have minimum or no internalization. Instead, once these agents bind to their cognate receptor, their function is blocked. The process of internalization of receptor agonists is the basis for an efficient accumulation of radioligands into the cell, which has been considered a crucial step in targeted tumor therapy with radiolabeled peptides. 16,18 From this consideration, agonists seem more potent candidates than the corresponding receptor antagonists for PRRT. However, recent studies on somatostatin receptor (SSTR)-targeting peptides have demonstrated that despite the minimal internalization of the receptor–antagonist complex into tumor cells, the antagonists bind to the receptor with higher affinity than the corresponding agonists and therefore have higher tumor uptake than agonists. 18,19 A reason for this observation may be that antagonists bind to a larger population of binding sites and have a lower dissociation rate than agonists. 18,19 Confirming data were also obtained with the gastrin-releasing peptide (GRP) hormone receptor. 20 A detailed discussion on recent developments of radiolabeled SSTR antagonists is provided in the PRRT for neuroendocrine tumors section of this review.
Radiolabeled Peptides for Targeted Therapy
Peptide-based agents for targeted radiotherapy comprise a regulatory peptide or its suitable synthetic analog (targeting vector), pharmacokinetic modifier (hydrophilic aliphatic or amino acid tethering molecule), BFCA (a complexing agent capable of producing a radiometal–ligand complex, which is kinetically inert in vivo, while linking it to the regulatory peptide), and a therapeutic radionuclide (Fig. 1).

Advantages and limitations of radiolabeled peptides as therapeutic agents
Advantages
The attractive features of radiolabeled small peptides over other radiolabeled biologically active molecules in therapy are as follows
11
: • Biologically active peptides can be synthesized without much difficulty by using either an automated peptide synthesizer or by manual synthesis. Automated synthesis is used for rapid production of simple peptides, whereas manual synthesis is ideal for long or difficult sequences, and peptide modifications. Known sequences of amino acids can be modified to slow their in vivo catabolic rate. • Possibility to radiolabel with a variety of radionuclides by using both conventional and novel chelating moieties. • Offer the feasibility of kit formulation strategy for the preparation of therapeutic agents. • Amenable to extensive chemical/molecular modifications to optimize their affinity for a particular receptor and to display a more specific biodistribution pattern. • Ability to tolerate the reaction conditions (pH, temperature, etc.) essential for carrying out necessary chemical modifications and radiolabeling. • Possibility to attach a chelating agent at the C- or N-terminus as well as to the free amine or carboxylic acid present on the peptide.
Limitations
• Criteria for successful therapeutic use of radiolabeled peptides are high target specificity, high binding affinity, long metabolic stability, and high target-to-background ratio. A short half-life in the circulation can be a major road block for its successful in vivo application owing to peptide susceptibility to degradation before reaching the intended target. To circumvent such enzymatic destruction, most peptides need to be synthetically modified. Considerable research efforts have been directed toward the development of metabolically stable peptides suitable for clinical use by carrying appropriate molecular modifications, such as the use of more stable
• The loss of binding affinity to the receptor and alteration in in vivo metabolism upon coupling with a chelator and/or introduction of a radiolabel are other potential challenges associated with the use of small peptides. This can often be effectively circumvented by inserting a spacer group between the targeting vector group (peptide) and the radioisotope-binding domain (Fig. 1) located between the binding sequence and the chelating moiety. 7 An appealing approach is the cyclization of the peptide around a metal core (i.e., often the ionic form of the radioisotope), which not only makes the peptide analogs resistant to chemical and proteolytic degradation in vivo but also can generally greatly increase the affinity for its target. 21,22
• Another challenge often associated with the use of radiolabeled peptides is the high kidney uptake and retention often observed, which is a concern because of potential renal damage, particularly for radionuclide therapy. 23 –25
Development of peptide-based therapeutic radiopharmaceutical
The major steps involved in the development of radiolabeled peptide for therapy are as follows
7,13
: (1) At the beginning, it is essential to identify the molecular target (receptor) with relevance to human disease. In this context, it is critical to identify not only the potential peptide receptors that may be of interest but also the tumor types that are particularly suitable to be targeted with a given peptide. For successful therapy, it is essential to have a thorough knowledge of the density of a peptide receptor in a given tumor type. This can often be determined in a personalized approach by tracer administration and imaging of receptor density using a peptide analog radiolabeled with a γ-emitting radioisotope. (2) Identification of a peptide, either natural or synthetic, that displays high affinity for the corresponding receptor system. The phage display library technique and the one-bead one-compound combinatorial library method are the two approaches that have been widely used for identification of a suitable peptide.
26
(3) Design and molecularly engineered synthesis of the peptide, followed by attachment of a BFCA through a metabolically resistant covalent bond. The BFCA is covalently attached to the peptide either directly or through a linker and strongly coordinates the radionuclide. The choice of the BFC is largely determined by the nature and oxidation state of the radiometal. An ideal BFC is able to form a stable radiometal chelate with high thermodynamic stability and kinetic inertness. Moreover, the BFCA should have minimal bulk so that the biodistribution and receptor-binding affinity of the parent peptide is not affected to a significant extent. (4) Selection of a suitable radionuclide with high radiotoxcity.
3
(5) Adaptation of a radiolabeling procedure that permits high labeling efficiency and high specific activity (SA). It is crucial to choose a radiolabeling procedure that retains the receptor-binding affinity of the peptide. (6) In vitro characterization must be assessed, such as the binding of a radiopeptide with tumor cells, stability of radiolabeled peptide in serum, receptor-binding affinity, internalization into the tumor cells, and dissociation from the tumor cells. The above characteristics are critical to recognize the potential peptide receptors and tumor types that are to be targeted with a given peptide. (7) In vivo evaluation of the radiopeptide with respect to tumor-targeting properties and in vivo stability using animal models. Preclinical evaluation of radiolabeled peptides is carried out by biodistribution and imaging studies in an appropriate animal model, preferably nude mice xenografts, which ensures the efficacy of the radiolabeled peptide for the particular receptor target in vivo. (8) After completion of all the preclinical tests, including toxicological studies in animal models, dosimetry data are obtained for finalizing the dose required for patient injection. These studies are performed by administration of diagnostically relevant doses of the radiopeptide to a small group of patients under appropriately approved protocols (Prephase I or Phase 0). These studies are then followed by Phase I/II/III clinical trials on a large population of patients subject to satisfactory clinical outcome.
These steps are schematically represented in Figure 2.
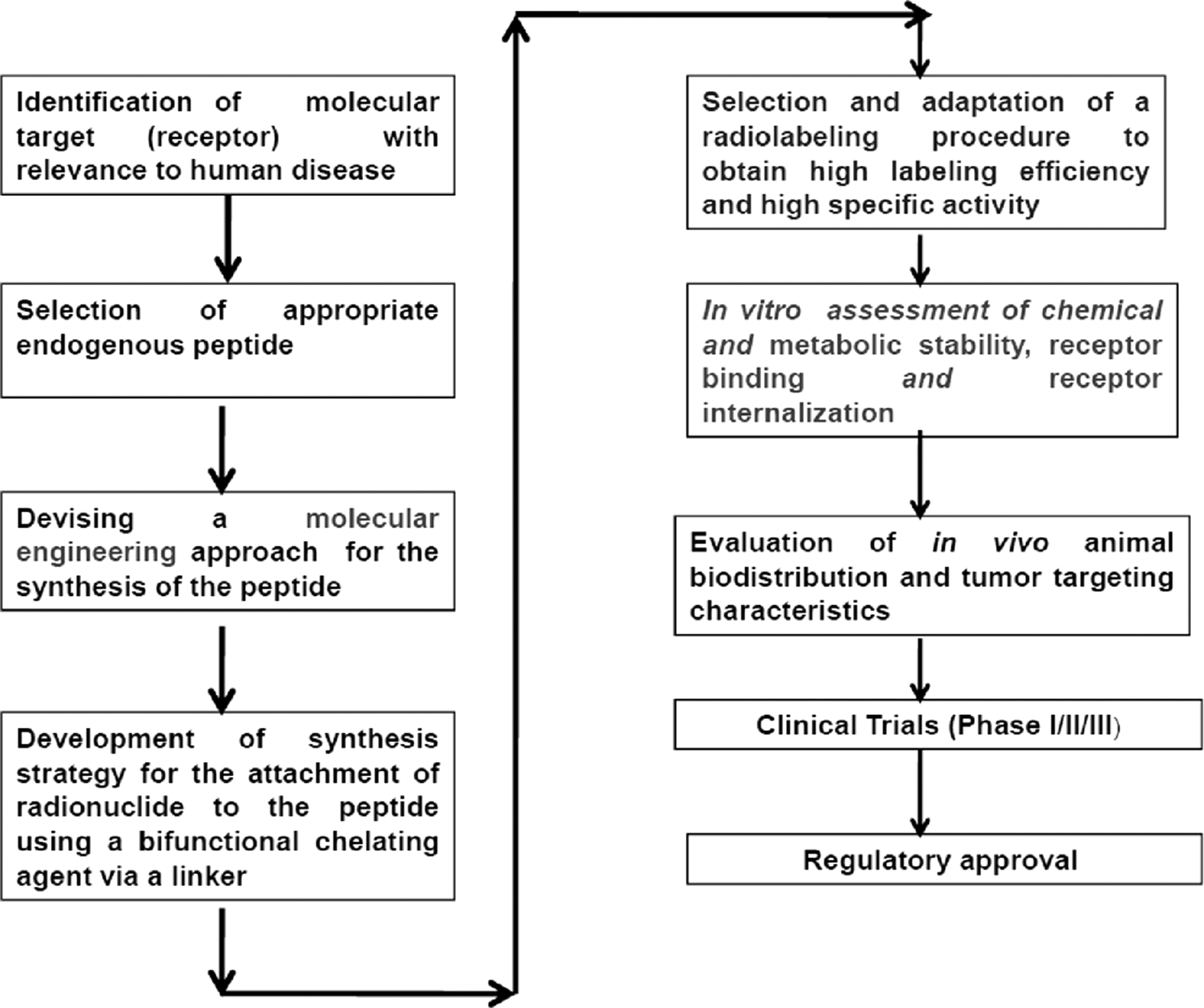
Major steps involved in the development of a peptide-based radiopharmaceutical.
Radionuclide selection
The inherent determinant for the success of PRRT resides in the selection of the appropriate radionuclide, which is based on a number of considerations described as follows: • Particle-emitting radionuclides that decay by β-particle emission, α-particle emission, and radionuclides that decay by electron capture and internal conversion leading to the emission of Auger and Coster-Kronig electrons are suitable for radionuclide therapy owing to their high linear energy transfer (LET) to deliver localized cytotoxic ionizing radiation. • The choice of emission type depends on the size of the tumor to be treated, intratumor distribution (i.e., degree of heterogeneity of radionuclide deposition), pharmacokinetics, and other factors. The physical half-life of the radionuclide should be matched well with the in vivo pharmacokinetics of the radiolabeled peptide. • The SA of the radionuclide preparation is an important criterion for PPRT utility. In light of the perceived need to induce pharmacologic effects at relatively low concentrations for PRRT, high SA radionuclidic preparations are mandatory. • While the radionuclide particulate emission properties determine the therapeutic potential, the presence of a γ-photon of 100–200 keV with low abundance can usually be very advantageous for low-dose imaging studies for initial personalized targeting evaluation, dosimetry estimates, evaluation of excretion, and for monitoring response to therapy. This is becoming an important attribute in the emerging field of personalized medicine.
27
• The radionuclide should have chemistry amenable to its attachment with a BFC, and binding must exhibit high in vivo stability when attached to the peptide.
Although one of the most important therapeutic radioisotopes used for several decades, the use of 131I in PRRT applications has not in general been particularly successful. Bakker et al. reported extensive radiolytic decomposition of octreotide for preparation of therapeutic doses of 131I-labeled octreotide for SSTR-targeted therapy, 28 possibly attributable to its radionuclidic decay properties. Yttrium-90 and 177Lu are the most successfully used β− emitting radionuclides, whereas the Auger electron-emitting radionuclide, 111In, is also explored, although with relatively less success for PRRT. Very recently, α-particle emitters such as 213Bi have been introduced for PRRT. 29,30 The characteristics of these and other useful radionuclides for PRRT are depicted in Table 3.
Production of radionuclides currently used in PRRT
Any advancement in PRRT will be largely dependent upon the availability of required quantity and quality of high SA radionuclides. The major challenge for the production of a radionuclide requires selection of the most economical process from a large pool of available options. Even though a radionuclide possesses attractive properties, the lack of an economical production process constitutes a major barrier for its utility in PRRT. To sustain the success of PRRT, it is important to assure access to appropriate production as well as radionuclide separation and purification technologies that will provide high SA radionuclides of desired quantities and qualities. Consequently, this has been an area of research and the production aspects of the three most current widely used radionuclides for PRRT are described below.
Indium-111
Indium-111 is produced commercially by irradiating a natural cadmium target with high-energy protons in a cyclotron according to the 111Cd(p,n)111In or 112Cd(p,2n)111In reaction, and both remain the most widely used 111In production methods. 31 –33 When production of 111In is carried out by bombarding the Cd target with protons, the 111In activity at the end of bombardment (EOB) contains radionuclidic contaminations, such as 109In (t½=4.3 hours), 110mIn (t½=4.9 hours), and 114mIn (t½=49 days). The first two radionuclides of indium have a relatively short half-life and hence cooling for 24 hours after EOB will reduce their contaminations. Alternatively, 111In is also produced by irradiation of a natural silver target with an energetic α-beam in a cyclotron according to the 109Ag(α,2n)111In reaction. 33 –36
Radiochemical separation of 111In from the irradiated target can be performed using a large variety of techniques, such as coprecipitation with Fe(OH)3 or La(OH)3, ion exchange, and extraction chromatography. Each of the techniques has its own advantages and disadvantages. Liquid–liquid extraction and ion-exchange chromatography (IEC) are widely used for radiochemical separation of 111In on the commercial scale. 31,32,35 –39 However, despite the development of robust separation and purification technology, low production yields are a major impediment in the large-scale production of 111In required for its therapeutic use.
Lutetium-177
Two alternative production routes are available to produce 177Lu with adequate SA for PRRT. The first is the direct route neutron activation of highly enriched 176Lu targets by the 176Lu (n,γ)177Lu reaction in medium to high flux reactors. 40 –48 The indirect route is based on neutron irradiation of ytterbium targets, by the 176Yb(n,γ)177Yb process, with subsequent β-decay to no-carrier-added (NCA) 177Lu. 49 –53 The radiochemical processing in the former case involves simple dissolution of the irradiated targets, whereas the latter route involves elaborate radiochemical processing to separate 177Lu from ytterbium targets as well as its radionuclides.
In contrast to most other (n,γ) methods of production, the direct neutron activation method of 177Lu production is attractive owing to the high thermal neutron capture cross section of 176Lu (σ=2090 b). It is pertinent to point out that the cross section of this reaction is the highest encountered among all (n,γ) produced radionuclides presently used for therapy. It is possible to produce 177Lu of SA of 740–1110 GBq/mg (20–30 Ci/mg) in a medium flux reactor using isotopically enriched 176Lu. During the process, 20%–30% of the 176Lu atoms are converted to 177Lu. The SA of 177Lu can be augmented to 1850–2405 GBq/mg (50–65 Ci/mg) by irradiation in high flux reactors, 40 such as the high flux research reactor at the Oak Ridge National Laboratory and the SM3 reactor in Dimitrograd, The Russian Federation. The (n,γ) method of 177Lu production described above also leads to the coproduction of the long-lived 177mLu impurity having a half-life of 160 days. A careful optimization of the time of irradiation is essential to obtain the highest SA as well as minimum contamination with 177mLu. 40,45,46,48
For access to NCA 177Lu, neutron irradiation of ytterbium targets, followed by radiochemical separation of 177Lu, has been evaluated by several groups. 49 –58 In this method, an isotopically enriched 176Yb target undergoes the (n,γ) reaction to produce 177Yb, which subsequently decays by β− emission (T½=1.9 hours) to yield 177Lu ( 176 Yb(n,γ) 177 Yb→ 177 Lu). The prospects associated with this route of 177Lu production along with the challenges associated with the separation of chemically similar neighboring lanthanides have thus led to considerable research and innovative radiochemical strategies. Different approaches have been explored in the separation of NCA grade 177Lu from large amounts of ytterbium. 49 –58 Successful preparative-scale separations of Lu from Yb for clinical applications have been reported in the literature by Lebedev et al., 53 Bilewicz et al., 54 Knapp et al., 55 Mirzadeh et al., 50 and Chakravarty et al. 49
Yttrium-90
NCA 90Y is required for the preparation of radiolabeled peptides used for targeted therapy since 90Y produced by (n,γ) activation in a nuclear reactor is of low SA due to the poor neutron absorption cross section (1.28 b) of 89Y. 59,60 NCA 90Y can be produced in a nuclear reactor by pursuing the 90Zr(n,p) 90 Y reaction using 100% enriched 90Zr target and fast neutron flux of ∼7.5×1013 cm2 s−1. 60,61 Although this method obviously holds promise as a viable approach, several issues related to the long-term availability and cost of enriched 90Zr, the requirement of a fast neutron flux, R and D associated with target design, and separation of 90Y and 90Zr recycling must all be resolved. The amount of 90Y produced by this route will also be limited because of a very low cross section for the 90Zr(n,p) 90 Y reaction. Separation of 90Y in the NCA form from 90Sr, making use of a radionuclide generator system, is the most appealing process to obtain large amounts of 90Y. 60 Strontium-90 is one of the major fission products of 235U with fission yield of 5.93% and can be isolated from aqueous nuclear fuel reprocessing high-level liquid waste solutions. 62 –68 Purity requirements for 90Sr for availing medically useful 90Y are very high and extensive quality controls are essential.
The separation of NCA 90Y from 90Sr is a challenging task owing to the necessity to maintain the levels of 90Sr contamination as low as possible for therapeutic applications of 90Y since ionic 90Sr localizes in the skeleton and owing to its long half-life has a very low maximum permissible body burden of 74 kBq (2 μCi) over patient lifetime. A variety of separation approaches based on solvent extraction, IEC, extraction chromatography, membrane-based separation, electrodeposition, etc., have been reported with an aim to isolate 90Y from 90Sr. 60,69 –78 A review of the 90Sr/90Y generator technologies indicates that an automated 90Sr/90Y generator based on an electrochemical separation 60,77 –79 could be efficiently adapted in centralized radiopharmacies set up for an assured supply of 90Y of required quality and quantity over a sustained period (>10 years). However, because of the very strict quality control requirements to eliminate 90Sr contamination from the product and the significant safety issues associated with the safe handling of high activity levels, most 90Y currently used is provided from a central commercial processing facility. Ensuring successful utilization of 90Y for targeted therapy demands quantitative estimation of Bq levels of 90Sr impurity in GBq quantities of 90Y and should be within the pharmacopeia-established limit. The reported extraction paper chromatography concept, 80 which combines chelate-based extraction with partition chromatography, is attractive as it would exploit the ability of 2-ethyl hexyl-2-ethyl hexyl phosphonic acid (KSM-17) to retain 90Y selectively at the point of spotting and at the same time offer the convenience of separating other impurities by partition chromatography.
Synthesis of peptides
The capabilities to undertake large-scale synthesis and modification of custom peptides to target specifically to tumors rather than to normal tissue are an important step and represent the cornerstone for the success of PRRT. Peptide synthesis is generally performed in a peptide synthesizer, which should have the ability to synthesize a broad spectrum of peptides. Liquid-phase peptide synthesis is used for very short dipeptide synthesis, while large-scale solid-phase peptide synthesis is well suited for preparing biologically active custom peptides in applications required for PRRT.
81
Postsynthesis purification of peptides obtained from any of these methods is, of course, a prerequisite. Before radiolabeling, the peptide is generally subjected to the following evaluations: • Characterization of the peptide by determining the amino acid sequence. • Determination of the peptide solubility in water at 25°C at neutral, low, and high pH, and (sometimes) in dimethylsulfoxide (DMSO), which can give valuable information for routine use in PRRT. • Purity of the peptide must be ensured. Typical specifications are 97%–99.9%. Impurities from peptide synthesis include deleted sequences due to incomplete coupling reactions, fragmented sequences, etc.
For the peptides to be suitable for administration in humans, care must be taken to ensure that the synthesis of the peptide is carried out according to good manufacturing practices (GMP), which guarantee total quality system for the following aspects of the synthesis: • The synthesizer needs to be in a controlled/validated condition. • Starting materials used for the synthesis must be held in quarantine and are required to be quality assured. • Use of standard operating procedures (SOPs) for the production and the purification peptides. • The analysis of the quality of the peptides should be part of the SOPs. • Synthesis and production of the peptide must be carried in an environmentally controlled area, including both the microbiologic quality and number of air-borne particles.
Bifunctional chelating agent
A BFCA usually contains three parts, which include a metal chelating, a spacer framework, and a conjugation group (Fig. 1). Between the targeting peptide and the radionuclide is the BFCA, one end of which is covalently attached to the peptide either directly or through a linker, while the other strongly coordinates to the metallic radionuclide. Selection of a BFCA is largely determined by the nature and oxidation state of the radiometal ion and requires an intimate understanding of the coordination chemistry of any given radiometal. The linker is often used for modifying the pharmacokinetic properties of the radiopharmaceutical. Depending upon the radionuclide and the BFCA, linker groups capable of rapid metabolism can increase the clearance of the radiolabeled peptide from the blood through the renal system.
The BFCA used for PRRT should possess the following criteria
82
–85
: • The conjugation group should be easily attached to the peptide. • Radiolabeling of the BFCA should be efficient and rapid when performed, preferably at room temperature and low concentration at a pH that is suitable for biological targeting vectors. • In case of macrocyclic chelators used as BFCAs, the cavity size of the chelator should be compatible with the ionic radius of the radionuclide such that all the donor groups can be properly aligned for optimal binding to the metal ion so as to provide high stability and limiting dissociation. • It is always preferable if the BFCA is able to withstand radiolytic decomposition because of high doses of β− radiation in the final preparation. However, there are several examples known for metal complexes, which have been stabilized using free radical scavengers and radioprotectants. • The BFCA must form a radiometal chelate with high thermodynamic stability and kinetic inertness at neutral pH to maintain the metal chelate intact under physiological conditions. Decomposition of the radiometal from the chelate can often release free radiometal ions, which may deposit on bones and result in bone marrow toxicity, particularly in the case of +3 metals such as 90Y and 177Lu. • The BFCA must form a metal chelate with a minimum number of spacial isomers. The tumor uptake of a radiolabeled peptide depends not only on the receptor-binding affinity of the peptide but also on pharmacokinetics, which is determined by the physical and chemical properties of both the biomolecule and the radiometal chelate. Formation of isomers may have a significant impact on the biological properties of the radiolabeled peptide and therefore must be carefully assessed. • The BFCA must have high hydrophilicity, which helps to improve the blood clearance and renal excretion of both the labeled and unlabeled BFCA–peptide conjugate. Rapid renal clearance of the unlabeled BFCA–peptide will minimize its competition with the radiolabeled BFCA–peptide for the receptor.
Common BFCAs utilized in PRRT can be grouped into two classes: acyclic (open chain) or macrocyclic (closed chain). There are a number of properties that can be used to validate acceptability of a BFCA in PRRT, including the following: (1) Thermodynamic stability constants, (2) Transchelation, (3) Acid catalyzed dissociation constants, and (4) Serum stability.
In recent years, a wide variety of acyclic and macrocyclic BFCAs (Fig. 3) have been developed from well-established chelating agents and used for the radiolabeling of small peptides.

Structure of acyclic and cyclic bifunctional chelating agents used for chelating radiometals.
Acyclic chelators
A significant advantage of acyclic BFCA is the more rapid metal-binding kinetics compared with macrocyclic complexes of comparable stability, resulting in faster radiolabeling, which can be a significant advantage for use of short-lived radionuclides. Unlike macrocyclic ligands, the acyclic chelators have the flexibility to form chelation rings that are dependent on metal-ion size and geometry. While a wide range of open-chain acyclic BFCAs have been developed and used, the promiscuity of these acyclic chelating agents that bind several ions in solution can be compromised by instability of the complex in vivo. In PRRT, the DTPA (diethylenetriaminepentaacetic acid) N3O5 chelating agent is the most commonly exploited acyclic bifunctional chelator. A major advantage of using DTPA analogs as BFCs is fast radiolabeling kinetics under mild conditions. 83,86 However, the kinetic liability of their metal chelates emerges as the major impediment when they are used as BFCAs for peptide and other biomolecules having slow blood clearance. 87
Macrocyclic chelators
Macrocyclic chelators usually offer the advantage over the corresponding acyclic chelators since the resulting radiometal complexes are more thermodynamically stable and remarkably more kinetically inert to dissociation (macrocyclic effect). DOTA-based macrocyclic chelators are the most widely used BFCAs in PRRT, and currently, widely used for the preparation of 90Y- and 177Lu-labeled peptides. 82,83,87 –90 The labeling kinetics of DOTA-based BFCAs are usually slow and dependent on the radiolabeling conditions, including the BFCA–peptide concentration, pH, reaction temperature and heating time, buffer agent and buffer concentration, and presence of trace metal ions as impurities. 83 However, the remarkable kinetic robustness of the DOTA complexes when subjected to a competing environment in the biological system makes DOTA-based BFCAs as the most ideal choice. 82,83,87 –90
Radiolabeling of peptides
Successful use of peptide-based agents for targeted delivery of therapeutic radionuclides involves development of appropriate radiolabeling procedures, which fulfill the following criteria: • There must be strong irreversible binding of the radionuclide with the peptide. • The procedure adapted should exhibit high radiolabeling efficiency and provide high SA of the radiolabeled peptide product. • The radiolabeling procedure should be simple, rapid, and reproducible. Formation of radiolabeled species should be fast at low temperatures, low concentration, and minimal excess of the peptides. A desirable radiometal should chelate with the peptide in solutions of nanomolar concentrations, preferably at room temperature within a few minutes. • Near-quantitative radiolabeling is desirable to avoid the necessities of postradiolabeling purification of the radiolabeled peptide. In most cases, this is difficult and a suitable purification step is thus essential and often requires microcolumn or HPLC purification, which is time-consuming and complex in a clinic-based radiopharmacy environment. • The radiolabeled peptides should be stable toward transchelation from potential donors present in vivo, hence kinetic inertness of the complex is the most important parameter. • The radiometal should ideally be selective for the BFCA–peptide conjugate. Particularly, complexation of metals, such as Ca2+, Cu+2, Fe+2, and Zn2+ ions, present in a high concentration in serum should be disfavored to avoid transmetallation in vivo.
• Biological activity of the radiolabeled peptide should be preserved during and after radiolabeling. • Important parameters to consider are the in vitro and in vivo stabilities of the radiolabeled species. Any decomposition or leaching of the radionuclide from the radiolabeled peptide should be within established acceptable limits.
Radiolabeling of peptides generally follows one of two approaches:
Direct radiolabeling method
The direct radiolabeling strategy without a BFCA is a relatively facile approach in the therapeutic arena predominantly used for the 188Re transition metal.
92
In this method, naturally occurring functional groups (i.e., -SH, -NH2, -OH, etc.) within the peptide sequence are used to complex to the radionuclide.
91,92
While this method offers a straightforward approach to radiolabel a peptide, it generally suffers from a number of the following limitations: • The lack of specificity of this radiolabeling strategy leads to the complexation of radiometals within or near the binding region of the biologically active peptide, which in turn inhibit the receptor-binding affinity. • The reduction of disulfide bonds (-S-S-→-SH) required to form the free SH-binding group requires the introduction of external reducing agents such as SnCl2, which can promote degradation of the peptide and result in lack of receptor binding. • While often successful using antibodies, the direct radiolabeling strategy has been observed to be ineffective for smaller peptides owing to the lack of reducible disulfide bonds necessary for metal chelation. Even in case reducible disulfide bonds are present in these smaller peptides, they are often a necessary component for effective biological activity for the maintenance of secondary and tertiary structural requirements.
Indirect radiolabeling method
To circumvent the problems often associated with the direct labeling method, the scope of introducing a BFCA moiety onto the biologically active compound at some geometric distance from the region necessary for receptor binding is most attractive. The BFCA forms a stable high-yield complex with the metallic nuclide, at the same time covalently linking the BFCA complex to the biologically active region of the peptide. 83 –86
In general, the two indirect radiolabeling approaches followed are either the prelabeling or the postlabeling method. In the postlabeling approach, a BFCA is first attached to the peptide either directly or through a linker to form the BFCA–peptide conjugate. The radiolabeling can be accomplished simply by reacting the BFCA–peptide with a radiometal salt solution in a buffer medium or, if necessary, in the presence of weak chelating agents (such as citrate). The postlabeling approach is of interest and utility particularly for biomolecules that are insensitive to the harsh experimental conditions sometimes encountered during the chelation process. Moreover, this approach is useful for the formulation of kits, where a convenient vial of the lyophilized vector is available for radiolabeling.
For heat sensitive peptides, the prelabeling approach seems to be an attractive alternative as it involves formation of the metal complex with a BFCA, followed by conjugation of the metal–BFCA complex, with the peptide in a separate step on the tracer level. In this approach, the chemistry is well defined, and the peptide is not exposed to the harsh experimental conditions typically encountered during the chelation step. While this approach obviously holds promise to perform the radiolabeling of heat sensitive peptides effectively, the requirement of a complex time-consuming radiolabeled procedure has emerged as the primary impediment that continues to thwart efforts for routine clinical use.
Present Status of PRRT Agents with Different Peptide Conjugates
In recent years, many radiolabeled peptide analogs have been developed or are at various stages of development for PRRT of different tumor types. These are somatostatin analogs, bombesin, vasoactive intestinal peptide (VIP), cholecystokinin/gastrin, neurotensin, and RGD derivatives, as summaried below.
Somatostatin receptor-specific peptides
Somatostatin receptors
SSTRs are, in general, membrane glycoproteins distributed in a variety of tissues throughout the body 94,95 and express specific binding affinity for somatostatin (SST). Somatostatin is a growth regulatory peptide, which inhibits secretion of hormones, and so can be used in the treatment of diseases caused by their overproduction. All SSTRs are not only linked to guanine nucleotide-binding proteins (G proteins) but also lead to inhibition of adenylyl cyclase following hormone binding 96 and are encoded by five genes, which are present on separate chromosomes. 97 There are five distinct human SSTR subtypes, SSTR 1–5, that have been identified and characterized. 12,98 The SSTRs are found on the cell membrane, and particular SSTR subtypes (SSTR 1–5) can occur alone or be grouped together in the same cell.
Human neuroendocrine-originated tumor cells often overexpress the somatostatin receptors, SSTR 1–5 (compared with the normal cells), with different intensity; while SSTR2 is by far the most abundant, SSTR4 is rarely expressed on tumor cells. A high density of SSTRs is found in neuroendocrine tumors (NETs), such as pituitary adenoma, pancreatic islet cell tumor, carcinoid, pheochromocytoma, paraganglioma, medullary thyroid cancer (MTC), and small-cell lung carcinoma. 99 The breadth of tumor cells, which overexpress these receptors, had been an important impetus, which lead to the development and use of therapeutic targeting vectors. Tumors of the nervous system, including meningioma, neuroblastoma, and medulloblastoma, also often express a high density of SSTR. Apart from these, tumors not known to originate from endocrine or neural cells may also express SSTR, such as lymphoma, breast cancer, renal cell cancer, hepatocellular carcinoma, prostate cancer, sarcoma, and gastric cancer. The expression of SSTR is not only specific for malignancy but also extended to certain benign lesions. For instance, active granulomas in sarcoidosis express SSTR on epithelioid cells and inflamed joints in active rheumatoid arthritis express them as well, preferentially within proliferating synovial vessels. 99 To develop radiolabeled agents for targeting tumors overexpressing SSTRs, a stable somatostatin analog peptide is used as the molecular carrier since the native somatostatin has a very short half-life in the circulation (2–3 minutes) owing to rapid enzymatic degradation. 100,101
In recent years, somatostatin analogs with high selectivity for each subtype of different SSTRs have been developed. Table 4 gives the affinity profiles of different somatostatin analogs and their BFCA conjugates. 102
SSTR, somatostatin receptor.
PRRT for neuroendocrine tumors
NETs represent a heterogeneous group of neoplasms, which originate mainly from the gastro-entero-pancreatic tract (GEP-NETs). The tumors include those arising from the endocrine cells within the respiratory and gastrointestinal tracts, known as carcinoid tumors. These have been classified as foregut, midgut, and hindgut according to their presumed embryological origins. They are characterized by their endocrine metabolism and histological patterns. As opposed to other tumor entities, these tumors range from well-differentiated slow-growing tumors to poorly differentiated highly invasive malignancies and may be functioning or nonfunctioning. NETs characteristically synthesize, store, and secrete a variety of peptides and neuroamines, which may lead to clinical syndromes, such as the carcinoid syndrome, the Zollinger-Ellison syndrome, or the Verner-Morrison syndrome. 103,104 However, most GEP-NETs are clinically silent until late presentation with metastases. Features such as neuroamine uptake mechanisms and/or specific receptors at the cell membrane such as SSTRs provide the scope for the identification, localization, and therapy of NETs. 105,106 Of the five major subtypes of SSTRs, which bind the 14-amino acid peptide somatostatin and its high-affinity 28-amino acid precursor, SSTR2 and SSTR5 are the ones that are most commonly expressed in NETs. However, there is considerable variation in SSTR subtype expression among the different tumor types and among tumors of the same type. 107 As the majority of NETs express SSTRs, they appear as an ideal target and provide scope for treatment with somatostatin analog (SST analog)-derived radiopeptides. 97,107 The extraordinary affinity of these peptides for SSTRs together with the internalization of the receptor–peptide complex facilitates retention of the radiopeptide in receptor-expressing tumors and at the same time their relatively small size facilitates rapid clearance from the blood. 108 Structures of several common DTPA and DOTA-coupled somatostatin analogs are depicted in Figure 4.
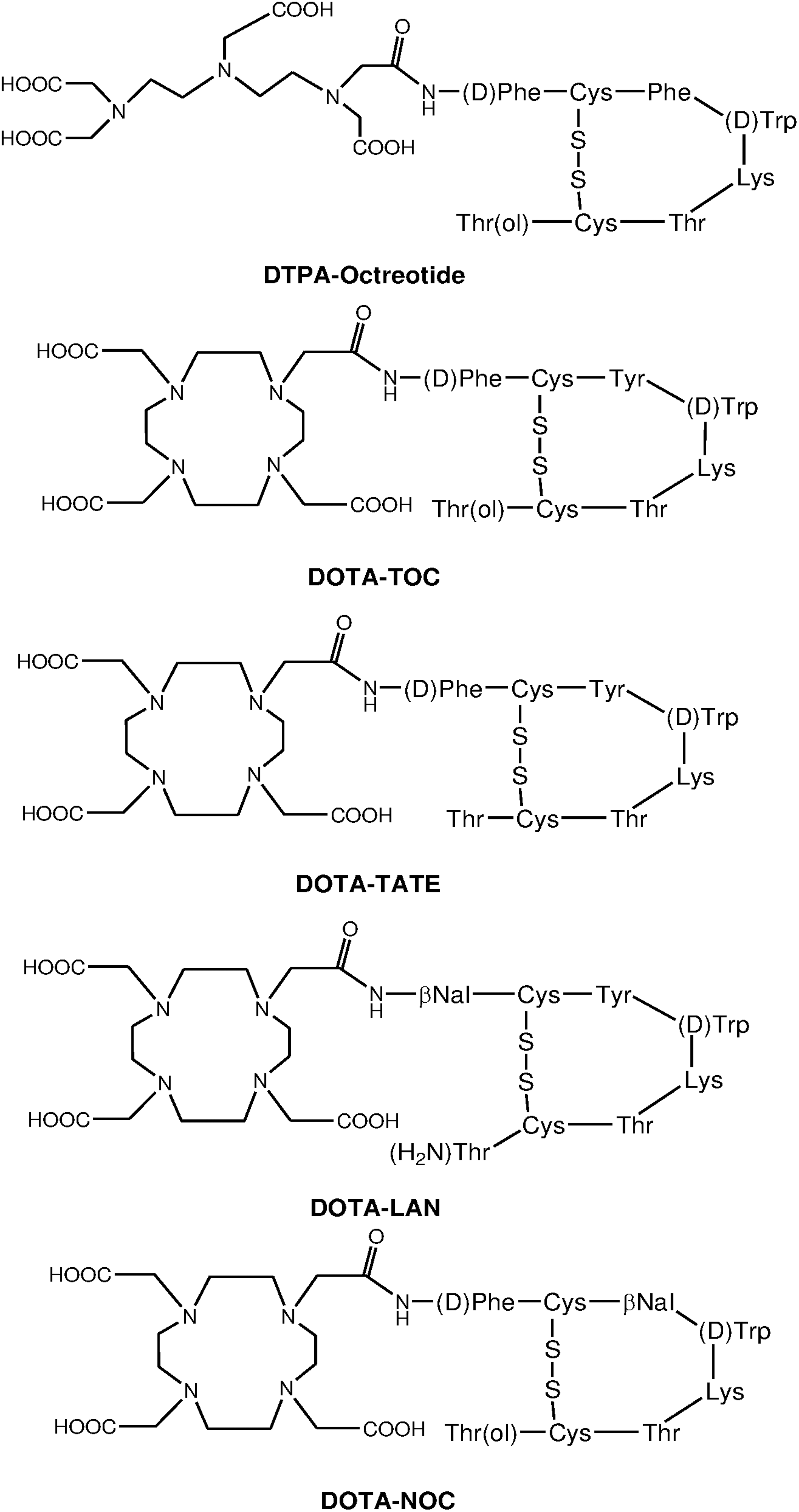
Structures of common DTPA/DOTA-coupled somatostatin analogs.
The most common SSTR-targeting radiopharmaceuticals are 111In-DTPA-octreotide, 90Y-DOTA0-Tyr3-octreotide (90Y-DOTATOC), 90Y-DOTA-Tyr3-octreotate (90Y-DOTATATE), and 177Lu-DOTA-Tyr3-octreotate (177Lu-DOTATATE). The DOTATATE derivative exhibits the highest affinity to SSTR2 and is hence widely used as a vector for targeted therapy. On the other hand, DOTA-lanreotide possesses lower affinity to SSTR2, although demonstrating considerable SSTR5 affinity.
102
Factors that determine the degree of uptake of radiolabeled SST analogs are as follows: (1) Stability of the radioligand. (2) Density of SSTR expression on the tumor. (3) Type of SSTRs expressed by the tumor. (4) Affinity of the radioligand for the SSTRs. (5) Efficiency of SSTR-mediated internalization. (6) Mass of the peptide administered.
Properties and use of the three most commonly used radiopharmaceuticals are discussed below.
111In-DTPA-octreotide: 111In-DTPA-octreotide was the first approved radiopharmaceutical for diagnostic imaging of SSTR-positive tumors. Investigators in the mid-to-late 1990s used 111In- DTPA-octreotide for PRRT, taking advantage of the Auger electron emission of 111In. The use of 111In-DTPA-octreotide in a high dose in patients with metastasized neuroendocrine tumors was often encouraging with regard to symptom relief, but tumor size regression was unsatisfactory. 109,110 The major roadblocks in the use of 111In-coupled peptides are the limited range of the Auger electrons and consequently short tissue penetration (∼10 μm) and very localized subsequent radiotoxicity. 111,112
Over the years, the targeting moiety of these peptides was modified by replacing Phe3 with Tyr3 in the octapeptide sequence (Fig. 4) to increase its hydrophilicity and to improve its affinity for SSTR2 compared with octreotide, and the DTPA chelator was replaced with DOTA to improve radionuclide–chelator complex stability. 113 Concurrently, 111In was replaced by more effective therapeutic radionuclides, such as 90Y and 177Lu, with sufficient β− energy to result in adequate cell damage. 81,101,111 –114
90Y-DOTA0-Tyr3-octreotide and 90Y-DOTA-Tyr3-octreotate: In an attempt to develop a more effective SST analog for PRRT, the modified [DOTA0,Tyr3]octreotide somatostatin analog was used in the second generation of SSTR-targeted radionuclide therapeutic agents, which have DOTA instead of DTPA as chelator. Such analogs not only possess a higher affinity for SSTR2 but also provide a more stable binding of β-emitting+3 metal radionuclides such as 90Y. In clinical studies conducted in different institutions with this SST peptide analog 90Y-DOTATOC or OctreoTher, the treatment outcome showed that 90Y-DOTATOC is effective for complete or partial remissions (10%–34%) in patients with NETs. 24,94,101,115,116 These ranges were higher than those obtained with 111In-DTPA0-octreotide. In light of the explicit need to develop a more effective SST analog for PRRT, [DOTA0,Tyr3]octreotate (DOTATATE) has been developed by replacing the C-terminal threoninol in DOTATOC with threonine. This small change of molecular structure (Fig. 4) was shown to have a ninefold higher affinity for the SSTR2 compared with DOTATOC in vitro. 102
While the use of 90Y-labeled peptides constitutes a successful paradigm of PRRT, a major drawback is the lack of γ-emission, which makes it difficult to obtain patient-specific dosimetry. Spurred by the need to estimate a specific dose on a tumor-specific personalized patient basis, the scope of imaging with analogs labeled with 111In or substituting the positron emitter 86Y for 90Y seemed to be a practical proposition to mirror the dosimetry and was followed diligently. It may also be mentioned that 90Y decays with very low abundance of positron emission, which also could be explored for imaging. 117 However, there are many practical advantages for use of a therapeutic radionuclide, which also decays with emission of photons for imaging.
177Lu-DOTA-Tyr3-octreotate: Lutetium-177, although reported more recently for use in targeted therapy, has already emerged as an important therapeutic radionuclide due to its more favorable radionuclidic characteristics as well as broad availability through straightforward production in high SA and high activity levels. 118 –122 Targeted radionuclide therapy with 177Lu-DOTA-Tyr3-octreotate (177Lu-DOTATATE, Lutathera®) has been extensively evaluated over a period of 10–15 years in major clinical centers in Europe and has demonstrated an overall response of 30%–38% and a significantly high median overall survival of 48 months. 23,112 –114,119 –122 It was observed that the quality of life improved significantly after treatment with 177Lu-DOTATATE. The Advanced Accelerator Applications (AAA) corporation acquired rights for marketing and sale of this new product with the aim of commercializing it in the United States as well as other parts of the world. Presently, Lutathera has received orphan drug status. In view of established effectiveness and safety, it is anticipated that the U.S. Food and Drug Administration (FDA) approval will be granted fairly swiftly. Presently, 177Lu-DOTATATE is in extensive use on an experimental basis or as a locally approved agent in several countries outside Europe, such as Brazil, India, and Uruguay. Figure 5 shows the post-therapy scintigraphic images of a typical patient presenting with extensive hepatic metastases recorded 2 hours after administration of 177Lu-DOTATATE along with 68Ga-DOTANOC PET scan of the same patient.
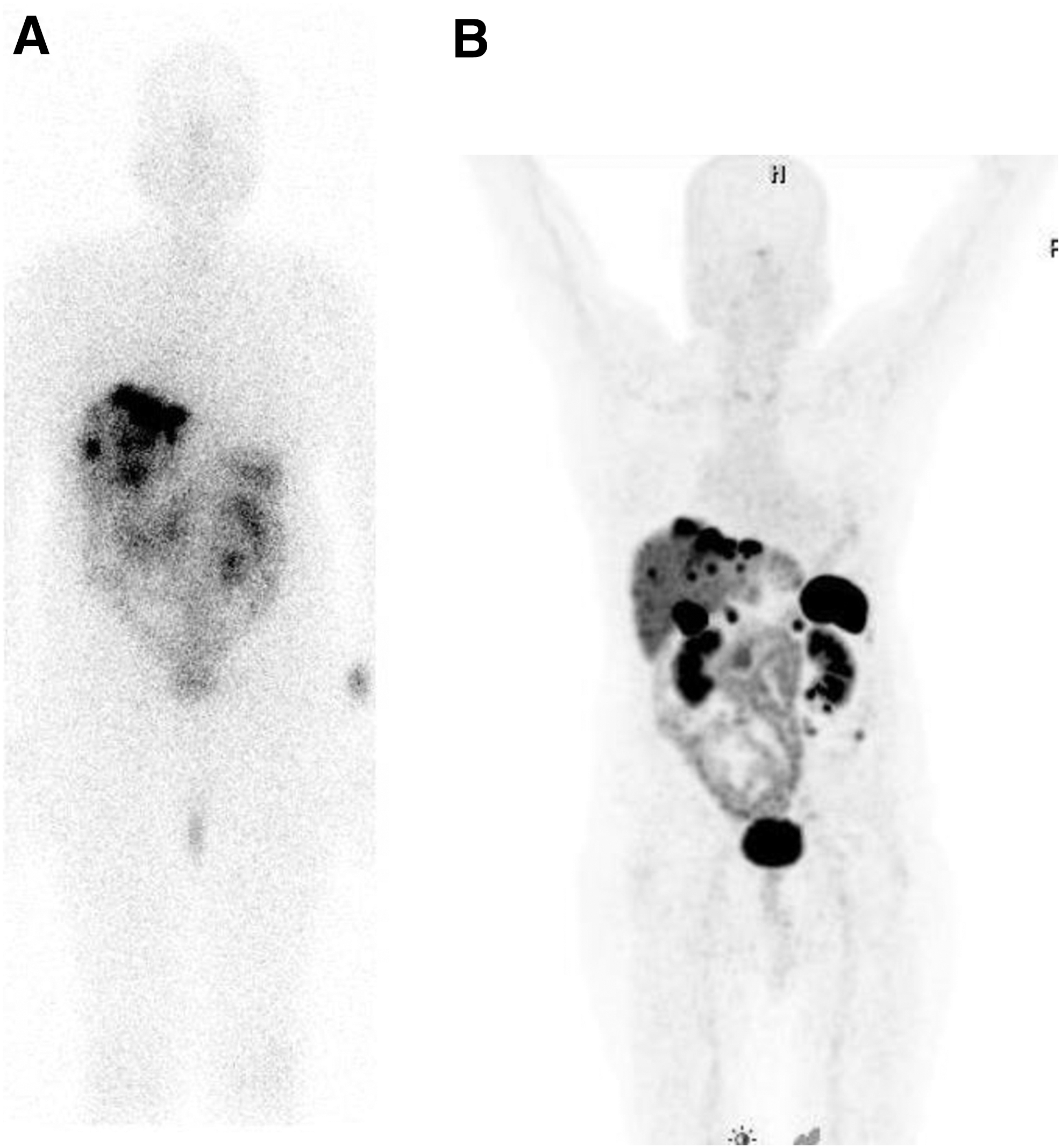
Animal experiments revealed that 90Y-labeled somatostatin analogs may be more effective for treatment of larger tumors, whereas 177Lu-labeled somatostatin analogs may be more effective in smaller primary tumors and micrometastases, but their combination may be even more effective. 122 In this context, the studies carried out by Kunikowska et al., 123 where a cocktail of 50% 90Y-DOTATATE+50% 177Lu-DOTATATE, activity wise, in 25 patients and comparison with another group of 25 patients treated by 90Y-DOTATATE alone, are important. The authors found striking differences in overall survival and progressive disease in favor of the combination 90Y+177Lu therapy compared with each single-agent treatment.
Radiolabeled SSTR antagonists: A very recent development in the field of tumor targeting with SSTR-specific peptides is the use of SST antagonists instead of agonists, such as octreotide, -TOC, and -TATE, which are presently in clinical use. A few preclinical studies have shown that tumor overexpressing SSTRs demonstrate higher uptake of radiolabeled SSTR antagonists than of SSTR agonists. 18,19,124 Wild et al. have demonstrated this phenomena using the 111In-labeled SSTR antagonist, DOTA-BASS (Fig. 6). 124 Cescato et al. have evaluated the in vitro binding behavior of the SSTR antagonist, 177Lu-DOTA-BASS, on several different well-characterized human tumor tissues by in vitro receptor autoradiography and compared the results with the established agonist radioligand, 177Lu-DOTATATE. 19 It was found that in all cases, the radiolabeled antagonist bound to more receptor sites than the agonist. The mean ratios of the uptake of antagonist, 177Lu-DOTA-BASS, to the agonist, 177Lu-DOTA-TATE, were 4.2±0.5 in the 9 ileal carcinoids, 12±3 in the 10 pheochromocytomas, 11±4 in the 7 breast carcinomas, 5.1±0.6 in the 10 renal cell carcinomas, and 4.8±0.7 in the 12 non-Hodgkin lymphomas.
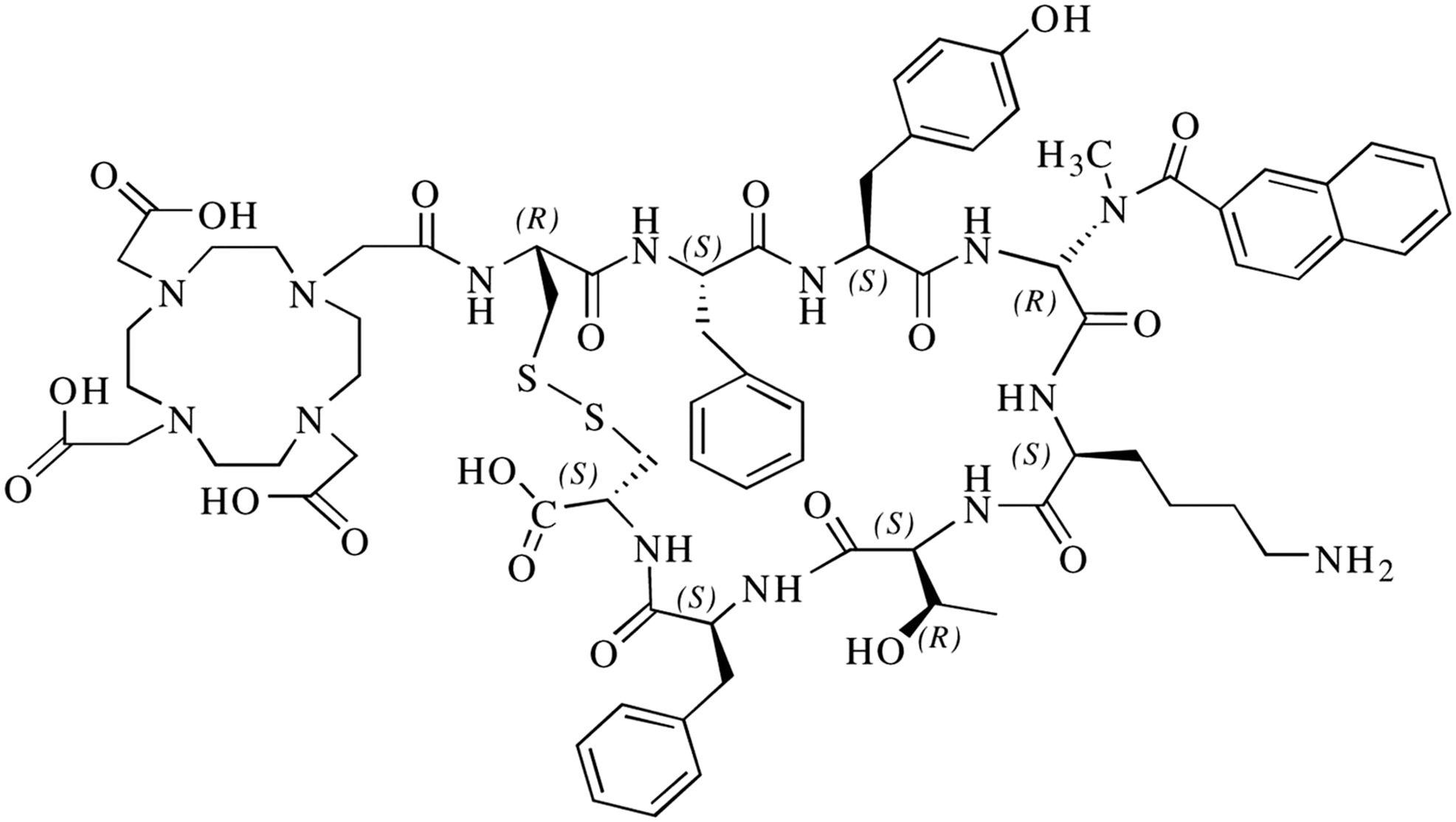
Structure of the somatostatin receptor (SSTR) antagonist DOTA-BASS.
Bombesin peptide analogs
Bombesin (BN) is a member of a family of brain-gut peptides, which play an important role in cancer. It is a 14-amino acid peptide present in amphibian tissues, a homolog of the 27-amino acid mammalian GRP present in humans. GRP and bombesin differ by only one of the 10 carboxy-terminal residues. Owing to their structural similarities, the biological activities of the two peptides are identical. GRP acts mainly in the central and enteric nervous systems and is primarily responsible for the regulation of several physiological processes, including thermoregulation, circadian rhythm, smooth muscle contraction, immune function, as well as the release of other peptide hormones. 125,126 Both BN and GRP exhibit high-affinity binding to the human GRP receptor, which is overexpressed by a variety of cancers, including prostate, breast, pancreas, gastrointestinal, and small-cell lung cancer. 10,127,128 Bombesin and GRP mediate their actions through membrane-bound G protein-coupled receptors, which include at least four different subtypes, namely the neuromedin B receptor subtype (BB1), the GRP receptor subtype (BB2), and the BB3 and BB4 subtypes. 10,127,128 Among these, GRP receptor (BB2) is of particular interest, which has been found to be overexpressed in a variety of tumors, including the lung, breast, GI, brain, and prostate, 10,127,128 and hence constitutes an attractive target for the detection and treatment of these cancers. Like other naturally occurring peptides, BN has a very short circulation half-life (<2 minutes). Radiolabeled BN-like peptides, which bind to BN/GRP receptors with high affinity and specificity, have emerged as options for site-directed radionuclide therapy. Various BN analogs based on their key amino acid sequence have been screened by incorporating radionuclides at the N-terminus of the peptide for therapy. One major impediment on the use of BN-like peptides is the tendency of the radiolabeled peptide to accumulate in the liver and intestines owing to high hepatobiliary clearance, despite their hydrophilic nature. 91 The presence of the GRP receptor in gastrointestinal tissues might be responsible to account for this high hepatobiliary clearance. 129 Several BN peptide analogs radiolabeled with 111In and 177Lu have been prepared and investigated for targeting of BN receptor-expressing tumors, including prostate and breast cancer. 130 –133 Cescato et al. have reported that radiolabeled GRP receptor antagonist N4-[D-Phe6,Leu-NHEt13,des-Met14]bombesin (6–14) (Demobesin 1) showed superior tumor-targeting capability in conjunction with less side-effects compared with GRP receptor agonist. 20 Such an observation indicates a paradigm shift in the field of PRRT using BN-like peptides.
VIP analogs
VIP is a 28-amino acid neuropeptide, isolated from the small porcine intestine. It is an important immunomodulator and stimulates the secretion of various hormones. In addition to its vasodilatory properties, it promotes growth and proliferation of normal and cancer cells mediated by cell surface receptors. 7,10,134 VIP receptors are found not only in the brain but also ubiquitously in majority of the human cell membranes of normal intestinal and epithelial cells. They are overexpressed on various tumor cells, such as colonic adenocarcinomas, pancreatic carcinomas, and carcinoids. The actions of VIP are mediated by two receptor subtypes (VPAC1 and VPAC2). While each of these receptor subtypes has different pharmacology and distributions, both of them have high affinity for VIP receptor subtypes. 135 In most of these tissues, the VIP receptor subtype preferentially expressed is the VPAC1 receptor, for instance, in hepatocytes, gastrointestinal mucosa, lobules and ducts of the breast, thyroid follicles, prostatic glands, urothelium of bladder and ureter, and acini of the lung and pancreatic ducts. One of the impediments on the use of VIP is its pharmacological toxicity, which necessitates an efficient purification step for the radiolabeled peptide from unlabeled cold peptide before administration. In vivo data using 123I-VIP as a universal ligand targeting VPAC1 and VPAC2 are available as proof of concept that VIP receptor-positive tumors, specifically gastrointestinal cancers, can be targeted in vivo in selected patients. 136,137 While the overexpression of VIP receptor on various tumor cells obviously holds promise as a treatment approach, the overexpression of VIP receptors on normal tissues, particularly in the lungs, central nervous system, liver, and intestine, limits the widespread therapeutic application of radiolabeled VIP analogs. Further efforts are thus required to assess the therapeutic potential of VIP analogs.
Cholecystokinin/gastrin peptide analogs
Cholecystokinin (CCK) and gastrin are structurally and functionally related peptide hormones that primarily function in the gastrointestinal tract and central nervous system. CCK and gastrin share an identical sequence of five amino acids at their biologically active C-terminal region. The biological action of these peptide hormones is mediated by CCK/gastrin receptors belonging to the superfamily of the G protein-coupled receptor. These receptors are distributed in a number of different tissues rather than as a selective expression of specific subtypes. They exert their functions through interaction with two receptors, CCK2/gastrin and CCK1 receptors. 138 CCK2/gastrin receptors are overexpressed in a high percentage (>90%) of MTC and in other tumors of small-cell lung cancers, astrocytomas, stromal ovarian tumors, and gastroenteropancreatic cancers. 139 Development of radiolabeled CCK/gastrin peptide analogs is beneficial for targeting the CCK/gastrin-expressing tumors in vivo. Recently, several radiolabeled CCK/gastrin peptide analogs have been prepared and evaluated for diagnostic imaging or radionuclide therapy of CCK/gastrin receptor-expressing tumors. 140,141 One of the main limiting factors for the development of PRRT using CCK2 receptors may be the high problematic kidney uptake with current CCK analogs. 142 To circumvent such a drawback, a new generation of CCK analogs has been designed with much less kidney uptake. 11,143
Neurotensin peptide analogs
Neurotensin (NT) is a 13-amino acid peptide found in the brain and gut and has many growth regulatory functions. NT is a neurotransmitter and a local hormone, and it can be found in the central nervous system and in peripheral tissues. Three NT receptor subtypes have been cloned to date, and overexpression of NT receptors has been found in many tumors. The biological effects of NT are mediated through the action of three different NT receptors, NTR1, NTR2, and NTR3, through the specific interaction of the peptide with cell surface receptors. 144 While NTR1 and NTR2 also belong to the superfamily of G protein-coupled receptors, the NTR3 is a single transmembrane domain receptor, which is similar to sortilin, a protein involved in receptor sorting. 145 Overexpression of NTR1 has been found in several human cancers, including Ewing sarcomas, meningiomas, astrocytomas, medulloblastomas, and pancreatic carcinomas. 139 While neurotensin peptide analogs hold promise as a treatment option, the initial clinical findings are not very favorable due to the high nonspecific uptake of radioactivity in the intestinal region and in the kidneys. 146
Glucagon-like peptide analogs
The Glucagon-like peptide receptor (GLP-1R) is also a member of the G protein-coupled receptor family and is secreted from L-cells in the gastrointestinal tract. 147 Glucagon is the principal counter-regulatory hormone that opposes insulin action, leading to coordinate bihormonal control of glucose homeostasis. Glucagon also inhibits glycogen synthesis and glycolysis in the liver. The biological active molecule of GLP-1 consists of 31 amino acids, derived from a precursor 36 amino acid sequence. The multifaceted physiological actions of GLP-1 include stimulation of the insulin gene expression, stimulation of insulin secretion, trophic effects on the β cells, inhibition of glucagon secretion, promotion of satiety, inhibition of food intake, and the reduction of gastric emptying, all of which contribute to normalizing elevated glucose levels. 148,149 GLP-1R is mainly expressed in the β cells of the islets of the pancreas, intestine, lung, kidney, breast, and brainstem. GLP-1 is overexpressed in most human insulinomas, gastrinomas, and pheochromocytomas. 150,151 GLP-1 is strongly overexpressed in greater than 90% of insulinomas with a density reported to be more than twice that of SSTR2. GLP-1 receptor agonists have a short half-life of less than 2 minutes in vivo due to degradation by the dipeptidyl peptidase-IV and tendency to rapidly degrade in the blood. 152,153 GLP-l analogs, such as exendin-4 and exendin-3, have similar biological activity, but longer in vivo half-lives. Exendin-4 and Exendin-3 are two 39-amino acid natural peptides, which are metabolically resistant and share nearly 50% homology with the human GLP-1. They differ by only two amino acid residues near the amino terminus. While initial results of in vitro as well as in vivo studies carried out with radiolabeled GLP-1 receptor-targeting analogs are encouraging, 154 –160 no clinical study has been reported thus far. High kidney retention of exendin-4 analogs could be the major impediment to realize the scope of using this analog for therapeutic purposes. Nevertheless, when this high accumulation in the kidneys can be mitigated, high GLP-1 receptor expression on tumors, in combination with the favorable in vivo characteristics of the recent exendin-4 analogs, offers GLP-1 receptor-targeted PRRT an attractive strategy.
RGD peptides for targeting integrin αvβ3 expression
Angiogenesis and integrins
Angiogenesis is a physiological process involving the formation of new blood vessels from pre-existing vessels. When the tumor size attains ∼2 mm3 volume, the increased interstitial pressure within the tumor appears to hinder the diffusion of metabolites and nutrients necessary for tumor growth. As a consequence, a state of cellular hypoxia initiates along with the sprouting of new blood vessels from the established vasculature or angiogenesis to facilitate the supply of oxygen and nutrients to tumor cells to survive and proliferate. 161 –164 The hypoxic condition increases cellular hypoxia-inducible factor transcription, leading to upregulation of proangiogenic proteins, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), or tumor necrosis factor-α (TNF-α). 163
The process of tumor angiogenesis is associated with the participation of a range of adhesion molecules, such as integrin, cadherin, selectin, and immunoglobulin families. 164 Among cell adhesion molecules (CAM), members of the integrin family play vital roles during various cancer stages, such as malignant transformation, tumor growth and progression, invasion, and metastasis. 165 –170 Integrins function as cell adhesion receptors for extracellular matrix (ECM) proteins, immunoglobulin, growth factors, cytokines, and matrix-degrading proteases. 166,167,170 –173 Integrins are divalent heterodimeric membrane glycoproteins comprising noncovalently associated α- and β-subunits. 174,175 Eighteen α-subunits and 8 β-subunits can assemble into 24 different heterodimers. Each subunit comprises an extracellular domain, a single transmembrane region, and a cytoplasmic region. The combination of α- and β-subunits determines the ligand-binding specificity and signaling properties of a given integrin, which can be classified based on their properties or based on their subunit composition. 174,175 The cell–cell and cell–matrix adhesion processes through binding of integrins to their ligands play critical roles in cancer proliferation and metastases. 167,176 –178 Therefore, the integrins have been proposed as the molecular targets for the treatment of cancer, 176,179 –181 vascular thrombosis, 182,183 and other diseases. 184,185
An array of integrins is involved in angiogenesis, among which αvβ3 is most important owing to its strong involvement in the regulation of angiogenesis. The αvβ3 integrin, which binds to arginine-glycine-aspartic acid (RGD)-containing components of the interstitial matrix, is significantly upregulated in the activated endothelial cells of the neovasculature surrounding tumor cells. 167,168,170,186,187 In addition, integrin αvβ3 is also upregulated in the cell membranes of various cancer cell types, including glioblastoma, neuroblastoma, osteosarcoma, melanoma, breast cancer, and lung cancer. 186,187
RGD peptide-based radiotherapeutics targeting integrin αvβ3
The RGD sequence emerged as the basic unit for a variety of molecules designed for the preferential binding to αvβ3 integrin and other integrins.
186,188
The RGD tripeptide itself is limited in the in vivo use because of its short circulation half-life. Conformational restriction by ring closure of the peptides and further chemical modification, such as the use of

Structures of mono-, di-, and tetrameric RGD peptide derivatives, which could be used for radiolabeling with 90Y and 177Lu using DOTA as the bifunctional chelator.

Whole-body scintigraphic images of C57/BL6 mice bearing melanoma tumors recorded
Multimerization of the RGD motif significantly improved the tumor uptake as well as retention, but at the same time resulted in increased uptake in nontarget organs, mainly the liver, intestine, lung, and kidney. 187,191,194 –196,202 –206 As a result, no significant improvement in tumor/kidney or tumor/liver could be observed by changing the targeting biomolecule from dimer to tetramer. Therefore, an important aspect of the research on integrin αvβ3-targeted therapeutic radiopharmaceuticals is to improve the T/B ratios by modifying excretion kinetics of radiolabeled cyclic RGD peptides while enhancing their high tumor specificity and long tumor retention. Toward this, Liu et al. have recently reported 90Y-labeled dimeric RGD peptide derivative viz. 90Y-DOTA-PEG4-E[PEG4-c(RGDfK)]2 (Fig. 9), where PEG4 used as the PKM linker. The radiolabeled agent effectively inhibited the growth of U87MG tumors in a nude mouse model along with significantly low radiotoxicity in normal organs compared with the case where no PKM linker was used. 208 The improved performance could be attributed to the following two reasons:

Structures of DOTA-PEG4-E[PEG4-c(RGDfK)]2 (3P-RGD2) and its 90Y complex.
(1) PEG4 liners improved radiotracer excretion kinetics from noncancerous organs.
(2) 90Y-DOTA-PEG4-E[PEG4-c(RGDfK)]2 probably binds to two adjacent integrin αvβ3 receptors in a bivalent manner, whereas 90Y-DOTA-E[c(RGDfK)]2 is monovalent. PEG4 linkers increase the distance between two RGD motifs from 6 bonds in E[c(RGDfK)]2 to 38 bonds excluding side arms of K-residues, making the former radioconjugate bivalent. 196,208
Despite recent significant advancements in this, the translation of the preclinical results from bench to bedside has not yet emerged.
PRRT Using α-Emitters
The application of α-particle–emitting radionuclides coupled to tumor-homing peptides is a relatively new approach in PRRT. α-Particles are characterized by a high LET rate leading to extremely high cytotoxicity on the cellular level and a short range in tissue, reducing side-effects in normal tissues. A few reports based on the preclinical evaluation on animal models demonstrate the potency and limited toxicity of α-emitters labeled with SST analog–peptide conjugates. 29,213 –215 The primary challenges that the established 90Y- or 177Lu-labeled SST analogs face are renal toxicities and incomplete treatments, especially in radioresistant tumors leading to a high relapse rate. 216 One possible solution to these problems could be the use of high LET α-emitters. Among the several α-emitters considered for therapeutic use, 211At and 225Ac/213Bi have been reported for use in PRRT. 211At was recently evaluated for targeting SSTR-expressing D341 Med human medulloblastoma s.c. xenografts in a murine model. 213 211 At is attractive due to its short half-life (7.2 hours), but has notable limitations due to production challenges and the presence of the long-lived 207Bi daughter product. Furthermore, 211At requires onsite cyclotron production and target-processing facilities. Recent studies have shown the potential of 213Bi in PRRT using SST analog–peptide conjugate viz. DOTA-TOC. 29,215213Bi can be readily obtained from a 225Ac/213Bi radionuclide generator system 29,215,217 and is attached to carrier molecules for therapeutic use. In another approach, 225Ac itself is used for radiolabeling, which eventually decays to 213Bi with the emission of four α-particles. 214 Biodistribution studies have demonstrated that 225Ac-DOTATOC effectively accumulates in nude mice bearing AR42J rat pancreas NET xenografts. 214 In addition, the activity levels of 225Ac-DOTATOC used for these studies were shown to be nontoxic (12–20 kBq), reduced the growth of NETs, and showed improved efficacy compared with 177Lu-DOTA-TOC. 214 Preclinical studies carried out with 213Bi-DOTA-TOC showed significant antitumor effects with minimal treatment-related organ toxicity in a rat pancreatic tumor model. 29 No acute or chronic hematologic toxicities were observed. These studies conclude that 213Bi-DOTA-TOC is a promising therapeutic radiopharmaceutical for further evaluation. Another significant development in the field of PRRT using α-particles is the use of 213Bi-DOTA-[Thi8,Met(O2)11]-substance P in the treatment of critically located gliomas. A pilot clinical study with the radioligand reported by Cordier et al. in five patients by direct administration of the agent into the inoperable glioma tumors showed significant tumor shrinkage without any local or systemic toxicity. 30
Summary and Future Outlook
PRRT can unveil numerous exhilarating therapeutic options for a wide variety of malignancies. The enormous research and clinical interest in the field of PRRT is a reflection of the major clinical importance of this topic. With a sensible strategy and sustained determination coupled with numerous innovations in molecular biology, the long-heralded potential of PRRT is finally being realized. This review provides a holistic overview of PRRT ranging from fundamental research to clinical realization. The breadth of literature cited reveals that the PRRT modality is indeed rapidly evolving and requires even more future improvements to fully realize its potential in the clinical arena.
The success of radiolabeled peptides as biological radionuclide-targeting vectors is largely related to the overexpression of peptide receptors in human cancers. The promise of PRRT is expected to reinvigorate innovation for studies to identify new peptide receptors overexpressed in specific tumors and will challenge the ingenuity of scientists to identify new radiopeptides and their development for potential clinical use in the previously defined targets along with the clinical efforts to optimize peptide receptor targeting. Prerequisites for treatment success include demonstration of high tumor uptake relative to nontarget tissues on quantitative, diagnostic radionuclide imaging and stable hematological and biochemical function. So far, PRRT is a promising therapeutic option in the management of inoperable or metastasized NETs. In patients with metastasized neuroendocrine tumors for whom surgery is no longer an option, PRRT appears to be the most effective therapeutic strategy with limited side-effects. PRRT with 90Y- and 177Lu-labeled somatostatin analogs has resulted in symptomatic improvement, prolonged survival, and enhanced quality of life of NET patients. Comparing 90Y and 177Lu, it is established that 90Y is more effective for larger tumors, while with 177Lu fewer relapses occurred when treating smaller lesions. Side-effects of PRRT are generally few and mild if adequate kidney protective measures are taken and dose limits are respected. From the patients' perspective, PRRT has often been described as a walk in the park, because treatment is typically not associated with the discomfort and side-effects that are commonly encountered in conventional chemotherapy.
With further progress in molecular biology technology, it is expected that superior receptor targets will be identified on cancer cells and PRRT is poised to grow as the next generation of anticancer therapeutics. The literature shows that it is possible to increase receptor density on tumor cells by using different methods. In PRRT, this would enable the administration of higher therapeutic doses to tumors, which might lead to a higher patient cure rate. In light of the perceived need to obtain optimal radiotherapy for large and small tumoral lesions, combinations of radiolabeled peptides with different radionuclides and multireceptor targeting using a combination of peptides seem to be an appealing approach, possibly because of more homogenous receptor targeting. The presence of multiple peptide receptors in selected cancers can be the basis for multireceptor targeting using two or more radiolabeled peptides in parallel. The motivation of realizing such a concept thus remains the primary focus of investigators in both the preclinical and clinical arena. Clearly, success in this direction demands a detailed understanding of the underlying biochemistry, physiology, and application in synthetic chemistry and radiopharmaceutical chemistry.
The results with long-acting SST analogs indicate that this therapeutic modality has its place in the treatment of patients with SSTR-positive NETs for size reduction, improvement of quality of life, and overall prognosis, but this should really be viewed largely as a springboard to spur further development in PRRT. New challenges and opportunities are waiting in the traditional domain of somatostatin analogs as well as outside of it. The success of PRRT lies not only on the continued fueling of the somatostatin analog applications with new discoveries but also an evaluation of a diverse range of other small regulatory peptides to realize their potential peptides as targeting ligands, as their receptors are overexpressed in various human tumors.
Nevertheless, apart from evaluation of somatostatin analogs, interest in the use of other emerging and promising peptides together with their respective receptors has steadily grown and is expected to yield positive results. Although this area has been relatively less explored, applications of new peptide–receptor combinations are highly desirable and there are good reasons to hope for a bright future in coming years. The stretching of the ingenuity of researchers has resulted in the emergence of a variety of peptides and BFCAs far more quickly in recent years than they did over the past decade. With the identification of several other different classes of receptors, such as gastrin, bombesin, and αvβ3 integrin, together with new peptide-receptor combinations, the scope of PRRT could be significantly amplified.
Despite the huge potential of PRRT, timely availability of therapeutic radionuclides is one major challenge, which currently obstructs the path for their widespread progress. To traverse these obstacles, a constant and reliable supply of therapeutic radionuclides of the required quality in the desired quantities needs to be assured. This would not only ensure a sustained growth and future expansion of PRRT but is also anticipated to empower future developments. While 177Lu is in routine production, additional research efforts are warranted to undertake the widespread availability of 90Sr/90Y generator to obtain clinical grade 90Y on a routine basis.
While 90Y and 177Lu are the commonly used radionuclides in PRRT, several other radionuclides with different physical characteristics (including α-emitters) are under investigation 29,30,214,215,218,219 and one would expect use of these radionuclides to gain momentum in the future. The availability of therapeutic radionuclides having desirable nuclear characteristics at high SA together with chemical methods to attach them stably to the peptides has also improved tremendously in the recent past. With the appropriate combination of an optimally engineered targeting peptide and a suitable therapeutic radionuclide, the benefit of PRRT is expected to swell substantially in the future.
In recent years, the trend has been for implementation of cGMP regulation for therapeutic radiopharmaceutical preparations. In light of the need to achieve such quality objective, a comprehensively designed and correctly implemented quality assurance system is of utmost importance. In this context, greater use of automated radiolabeling modules will represent a step in the right direction as automation not only ensures reproducible and robust radiolabeling but will also comply with qualification and validation protocols established by regulations (GMP, cGMP, GMP-like, or local regulations). Such a strategy is expected to become one of the important strides for bridging the development and application of new radiolabeled receptor-avid peptides into clinical practice.
While PRRT has emerged as a promising treatment option and made considerable headway, a number of roadblocks that impede progress need to be surmounted to attain a significant foothold in radionuclide therapy. In spite of the spectacular progresses accomplished thus far, there are many preclinical research results reported in animal models, which have failed to translate into clinical reality, despite favorable in vivo biodistribution profiles in animal models of disease. While efforts in the direction have attracted a staggering degree of attention, they deserve further exploitation not only because a greater range of treatment options will be needed but also for the adaptability to treat diverse diseases. Various peptide-based radiopharmaceuticals have demonstrated good tumor uptake; however, they have frequently shown nonspecific uptake in normal organs, such as the liver and kidneys. In light of these premises, considerable R and D and further developments are warranted in the coming decade to establish their potential in PRRT. Adequate resources and multidisciplinary commitments are essential to investigate numerous unexplored possibilities and assure translation of some of these promising preclinical findings of PRRT from the bench to the bedside, ideally in the near future.
The exciting new possibilities in PRRT are just starting to be realized; however, despite the large body of evidence regarding efficacy and clinical safety, PRRT is still considered as an investigational treatment. With rapid advancement in genomic and proteomic research, it is possible to identify the presence of new peptide receptor types/subtypes for efficient tumor targeting for therapy. The development and application of PRRT has a long way to go till new agents continue to be developed and approved. However, the ability to insure rapid progress of PRRT will depend to a great extent on the integration of emerging molecular biological research, radiochemistry, radiation physics, radiopharmaceutical science, and biotechnology with medical science. It is the responsibility of all the stakeholders, including research scientists, commercial manufacturers, clinicians, and hospitals, to share a common platform to identify the roadblocks and find routes to harness the immense potential of PRRT for today and tomorrow.
Footnotes
Acknowledgments
The authors gratefully acknowledge Dr. K.G. Kallur of Bangalore Institute of Oncology, Bengaluru, India, for proving post-therapy images of 177Lu-DOTA-TATE recorded at his institute. The authors also express their thanks to Dr. H.D. Sarma, Radiation Biology and Health Sciences Division, Bhabha Atomic Research Centre, Mumbai, India, for providing scintigraphic images of 177Lu-DOTA-(RGD)2 in the tumor-bearing animal model.
Disclosure Statement
There are no existing financial conflicts.