
Abstract
Minigastrin radiotracers, such as [111In-DOTA]MG0 ([111In-DOTA-DGlu1]minigastrin), have been considered for diagnostic imaging and radionuclide therapy of CCK2R-positive human tumors, such as medullary thyroid carcinoma. However, the high kidney retention assigned to the pentaGlu2–6 repeat in the peptide sequence has compromised their clinical applicability. On the other hand, truncated des(Glu)2–6-analogs, such as [111In-DOTA]MG11 ([111In-DOTA-DGlu10,desGlu2–6]minigastrin), despite their low renal uptake, show poor bioavailability and tumor targeting. [111In]CP04 ([111In-DOTA-DGlu1–6]minigastrin) acquired by Glu2–6/DGlu2–6 substitution showed promising tumor-to-kidney ratios in rodents. In the present study, we compare the biological profiles of [111In]CP04, [111In-DOTA]MG11, and [111In-DOTA]MG0 during in situ neutral endopeptidase (NEP) inhibition, recently shown to improve the bioavailability of several peptide radiotracers. After coinjection of the NEP inhibitor, phosphoramidon (PA), the stability of [111In]CP04 and [111In-DOTA]MG0 in peripheral mouse blood increased, with an exceptional >14-fold improvement monitored for [111In-DOTA]MG11. In line with these findings, PA treatment increased the uptake of [111In]CP04 (8.5 ± 0.4%ID/g to 16.0 ± 2.3%ID/g) and [111In-DOTA]MG0 (11.9 ± 2.2%ID/g to 17.2 ± 0.9%ID/g) in A431-CCK2R(+) tumors at 4 hours postinjection, whereas the respective increase for [111In-DOTA]MG11 was >6-fold (2.5 ± 0.9%ID/g to 15.1 ± 1.7%ID/g). Interestingly, kidney uptake remained lowest for [111In-DOTA]MG11, but unfavorably increased by PA treatment for [111In-DOTA]MG0. Thus, overall, the most favorable in vivo profile was displayed by [111In-DOTA]MG11 during NEP inhibition, highlighting the need to validate this promising concept in the clinic.
Introduction
The overexpression of cholecystokinin subtype 2 receptors (CCK2Rs) has been demonstrated in several human tumors, including medullary thyroid carcinoma (MTC), small cell lung cancer, astrocytomas, and stromal ovarian cancers. 1 –3 This finding combined with the lack of CCK2R expression in healthy surrounding tissues provides promising opportunities for high-contrast diagnostic imaging and radionuclide therapy of tumors with CCK2R-seeking radioligands. 4 –11 Minigastrin (MG, Leu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2)-based radioligands developed for such purposes, such as [111In-DOTA]MG0 ([111In-DOTA-DGlu1]MG), showed high CCK2R affinity and good metabolic stability, resulting in high targeting of CCK2R-positive lesions in animals and in man. However, their clinical applicability has been largely restrained by unfavorably high retention in the kidneys. 12 –14
One approach to suppress renal accumulation has been the full truncation of the implicated pentaGlu2–6 chain in the minigastrin sequence. 15 Yet, this modification has inadvertently led to poor metabolic stability and suboptimal tumor localization of the corresponding radioligands, such as [111In-DOTA]MG11 ([111In-DOTA-DGlu1,desGlu2–6]MG). 10,11,16,17 Alternatively, a number of structural modifications have been directed to the critical pentaGlu2–6 repeat of the minigastrin motif, including substitution of selected or all five Glu residues by Gln, or DGlu, to modulate charge, charge distribution, and stereochemistry in this part of the molecule. 18,19 The radiotracer [111In]CP04 ([111In-DOTA-DGlu1–6]MG) was accordingly developed by complete Glu2–6/DGlu2–6 substitution within the frameworks of a concerted European COST Action (BM0607 Targeted Radionuclide Therapy). Owing to attractive tumor-to-kidney ratios achieved in animal models, [111In]CP04 was selected among 12 other CCK2R-targeting radiopeptide candidates for further clinical validation in MTC patients. 16
We have recently proposed an innovative approach to enhance the bioavailability and thereby the tumor localization of a wide array of peptide radioligands, including those based on minigastrin. 20 This strategy exploits the prominent role of neutral endopeptidase (NEP) in the in vivo catabolism of peptide radiotracers, relying both on its ubiquitous and abundant expression in various tissues and organs of the body, as well as on its broad substrate specificity. 21 The impact of NEP on metabolic stability has been often overseen in the field of nuclear medicine, primarily because NEP is an ectoenzyme anchored on vasculature walls and on the surface of tissue cells, but absent in serum or plasma typically used for measuring radiopeptide stability. Coinjection of a potent NEP inhibitor, such as phosphoramidon (PA), 22,23 together with a radiopeptide allows high supply of intact radioligand to receptor-expressing tumor sites, thereby inducing significant enhancement of tumor uptake.
In the present work, we have been interested to assess the efficacy of this strategy in three distinct minigastrin radiotracers differing in the pentaGlu2–6 chain, which is critical for radiopeptide metabolic stability, tumor localization, and renal retention. For this purpose, we have conducted a head-to-head comparison of [111In-DOTA]MG0, its DGlu2–6 counterpart [111In]CP04, and the truncated des(Glu)2–6 analog [111In-DOTA]MG11 during in vivo NEP inhibition by PA. Emphasis has been given on the assessment of potential benefits in overall pharmacokinetics of the three structurally different radiotracers provided by PA administration, especially regarding tumor-to-kidney ratio enhancement.
Materials and Methods
Compounds and instrumentation
All chemicals used were reagent grade. CP04, DOTA-MG0, and DOTA-MG11 (Fig. 1), as well as DG2 (Demogastrin 2, N4-Gly-DGlu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2; N4, 6-(carboxyl)-1,4,8,11-tetraazaundecane)
9
used for CCK2R blockade, were purchased from piCHEM. [Leu15]gastrin (pGlu-Gly-Pro-Trp-Leu-(Glu)5-Ala-Tyr-Gly-Trp-Leu-Asp-Phe-NH2) was obtained from Bachem. PA (N-(α-rhamnopyranosyloxyhydroxyphosphinyl)-
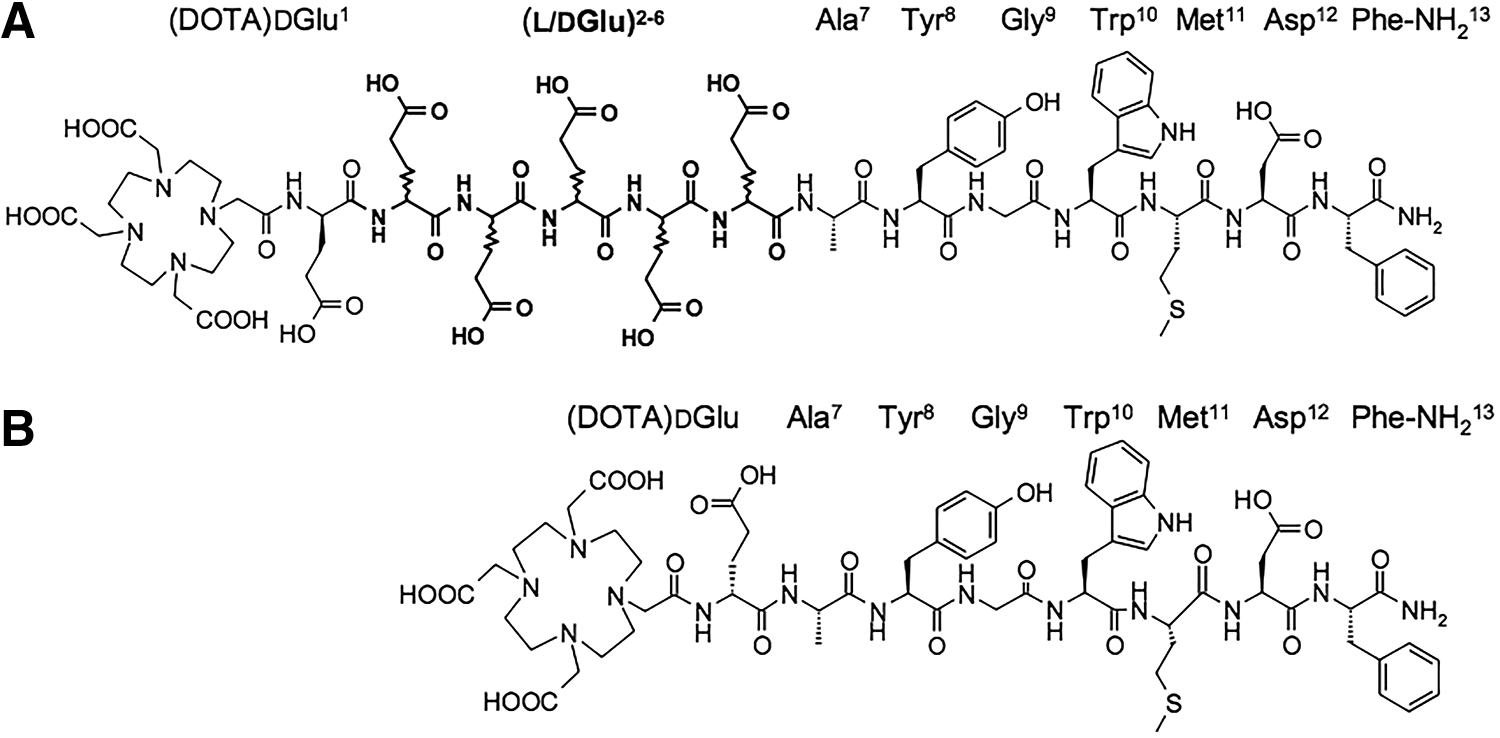
Chemical structure of DOTA-MG0 ([DOTA-DGlu1,LGlu2–6]MG), CP04 (([DOTA,DGlu1-6]MG)
High-performance liquid chromatography (HPLC) analyses were conducted on a Symmetry Shield RP18 column (3.9 × 20 mm, 5 μm) using Waters Chromatograph with a 600 solvent delivery system coupled to a 996 photodiode array UV detector and a Gabi gamma detector (Raytest RSM Analytische Instrumente GmbH). The Millennium Software was used for data processing and for controlling the HPLC system. For radioactivity measurements, an automated well-type γ counter [NaI(Tl) crystal, Canberra Packard Auto-Gamma 5000 series instrument] calibrated for 111In or 125I was used.
Radiolabeling and quality control
The lyophilized peptides were dissolved in water to a final concentration of 1 mM, and 50 μL aliquots were stored at −20°C. Labeling with 111In was conducted by adding 10 nmol peptide per 37–74 MBq of 111InCl3 in 0.1 M sodium acetate buffer and 0.1 M
Radioiodination of [Leu15]gastrin was performed following the chloramine-T methodology and [125I-Tyr12,Leu15]gastrin was isolated in high purity by HPLC. A stock solution in 0.5% bovine serum albumin in phosphate buffer saline (BSA-PBS) was kept at −20°C and aliquots thereof were used for competition binding assays (specific activity 2 Ci/μmol).
All manipulations with γ-emitting radionuclides and their solutions were performed behind suitable shielding in dedicated laboratories in compliance with national and international radiation safety guidelines.
Preparation of [natIn]CP04
Metalation of CP04 with natIn was conducted by adding 70 nmol peptide per 350 nmol natIn(NO3)3 in 0.1 M sodium acetate buffer (pH 3–5). To prevent oxidation of Met residue to the corresponding sulfoxides, 0.2 M
Cell culture
Human epidermoid carcinoma A431 cell lines transfected to stably express the human CCK2R (A431-CCK2R(+) cells) or mock transfected with an empty vector (A431-CCK2R(−) cells) were a generous gift from Prof. O. Boerman (Department of Nuclear Medicine, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands) and Prof. L. Aloj (Istituto di Biostrutture e Bioimmagini, Consiglio Nazionale delle Ricerche, Naples, Italy).
24
Cells were kept in controlled humidified air containing 5% CO2 at 37°C and were cultured in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX-I supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 4500 mg/L
Competition binding assays in A431-CCK2R(+) cell membranes
The binding affinity of CP04 and [natIn]CP04 for the human CCK2R was determined by competition binding experiments in A431-CCK2R(+) cell membranes harvested as previously described. 25 [125I-Tyr12,Leu15]gastrin served as competitive radioligand and [Leu15]gastrin as reference. Increasing concentrations of test peptide (10−5 to 10−13 M) were incubated for 1 hour at 22°C with the radioligand (50 pM, ∼30,000 cpm) and the membrane homogenate in a total volume of 300 μL binding buffer (BB, 50 mM HEPES pH 7.4, 1% BSA, 5.5 mM MgCl2, 35 μM bacitracin). Binding was interrupted by addition of ice-cold washing buffer (WB, 10 mM HEPES pH 7.4, 150 mM NaCl) and rapid filtration (Whatman GF/B filters presoaked in BB) on a Brandel Cell Harvester (Adi Hassel Ing. Büro). Filters were washed with ice-cold WB and were measured for radioactivity in the γ counter. IC50 values were calculated by nonlinear regression according to a one-site model applying the PRISM 2 program (Graph Pad Software). Results are expressed as mean ± SD of four experiments performed in triplicate.
Cell association and internalization in A431-CCK2R(+) cells
Radioligand cell association and internalization experiments were performed in A431-CCK2R(+) cells grown for 24 hours to confluency in six-well plates. On the day of the experiment, cells were washed twice with ice-cold internalization medium (DMEM Glutamax-I with 1% fetal bovine serum). Fresh medium (1.2 mL) was added to the cells, followed by [111In]CP04, [111In-DOTA]MG0, or [111In-DOTA]MG11 (150 μL, 250 fmol total peptide in 150 μL 0.5% BSA-PBS, 50,000–90,000 cpm); nonspecific values were determined by a parallel triplicate series containing 1 μM DG2. 9 Cells were incubated at 37°C for 60 minutes and incubation was interrupted by removal of the medium and rapid rinsing with ice-cold 0.5% BSA-PBS. Membrane-bound and internalized fractions were separated by 2 × 5 minutes of treatment in acid wash buffer (50 mM glycine in 0.1 M NaCl, pH 2.8). Cells were rinsed with 0.5% BSA-PBS, lysed with 1 N NaOH, and lysates were collected. Samples were measured for their radioactivity content in the γ-counter, and the percentage of internalized and membrane-bound fractions was calculated using the Microsoft Excel program. Results represent average value ± SD from 4 ([111In]CP04) or 3 ([111In-DOTA]MG0 and [111In-DOTA]MG11) experiments performed in triplicate.
Metabolic studies in mice
A bolus of [111In]CP04, [111In-DOTA]MG0, or [111In-DOTA]MG11 (100 μL, 11–22 MBq, 3 nmol total peptide in vehicle: saline/EtOH 9/1 v/v) was injected together with vehicle (100 μL; control group) or with PA (100 μL of vehicle containing 300 μg PA; PA group) in the tail vein of healthy Swiss albino mice (NCSR “Demokritos” Animal House Facility). Blood was withdrawn 5 minutes postinjection (pi) and directly placed in prechilled polypropylene tubes containing EDTA and
Biodistribution in SCID mice bearing A431-CCK2R(+/−) xenografts
Inocula (∼150 μL) containing a suspension of freshly harvested A431-CCK2R(+) cells (1.6 × 107) or A431-CCK2R(−) cells (1.4 × 107) in sterile physiological saline were subcutaneously injected in the flanks of 6-week-old male SCID mice (NCSR “Demokritos” Animal House Facility). After 6–8 days, well-palpable tumors (190 ± 80 mg) were grown at the inoculation sites and biodistribution was performed. A bolus of [111In]CP04, [111In-DOTA]MG0, or [111In-DOTA]MG11 (100 μL, 37–74 kBq, 10 pmol total peptide in saline/EtOH 9/1 v/v) was intravenously injected in the tail of mice together with vehicle (100 μL; control group) or with PA (100 μL of vehicle containing 300 μg PA; PA group). Animals were sacrificed in groups of five at 4 hours pi. Blood samples were collected and organs of interest as well as tumors were dissected, weighted, and counted in the γ counter; stomachs were emptied of their contents. Results were calculated as percent injected dose per gram tissue (%ID/g) with the aid of suitable standards of the injected dose using the Microsoft Excel program and are reported as mean ± SD.
All animal experiments were carried out in compliance with European and national regulations and after approval of protocols by national Authorities.
Statistical analysis
The unpaired two-tailed Student's t-test of Graph Pad Prism Software was used for statistical analysis; p-values of less than 0.05 were considered statistically significant.
Results
Radioligand preparation
Radiolabeling of CP04, DOTA-MG0, and DOTA-MG11 with 111InCl3 was completed by 10–15 minutes of incubation in acidic medium at 90°C in the presence of excess methionine to suppress sulfoxide formation and EDTA to bind any free 111In. More than 97% of radiometal was incorporated in the DOTA chelator of the peptides at specific activity of 3.7–7.4 MBq/nmol and >95% radiochemical purity was achieved, while recovery from the column was quantitative. These results allowed the use of the forming radioligands without purification in all subsequent experiments in EDTA and Met-containing solutions. Quality control before and after biodistribution experiments revealed the preservation of high radiotracer purity in the injectates till completion of the study.
Affinity of CP04 and [natIn]CP04 for CCK2R
The affinity of CP04 and [natIn]CP04 for the human CCK2R was determined by competition binding experiments in A431-CCK2R(+) cell membranes. As shown in Figure 2, both unmetalated and natIn-metalated CP04 displaced [125I-Tyr12,Leu15]gastrin from the human CCK2R binding sites in a monophasic and dose-dependent manner. The binding affinity of CP04 to CCK2R (IC50 = 1.28 ± 0.09 nM, n = 4) was found comparable with [Leu15]gastrin (IC50 = 0.86 ± 0.05 nM, n = 4), while incorporation of natIn led to a minor drop of receptor affinity (IC50 = 3.17 ± 0.21 nM, n = 4).

Displacement of [125I-Tyr12,Leu15]Gastrin from CCK2R binding sites in A431-CCK2R(+) cell membrane homogenates by increasing concentrations of CP04 (•, IC50 = 1.28 ± 0.09 nM), [natIn]CP04 (▲, IC50 = 3.17 ± 0.21 nM) or [Leu15]Gastrin (×, IC50 = 0.86 ± 0.05 nM, reference); results are presented as mean ± SD of four experiments performed in triplicate.
Radioligand internalization in A431-CCK2R(+) cells
As shown in Figure 3, [111In]CP04, [111In-DOTA]MG0, and [111In-DOTA]MG11 efficiently internalized in A431-CCK2R(+) cells after 1 hour of incubation at 37°C, showing a minor portion bound on the cell membrane, as expected for receptor radioagonists. This process was receptor specific, as suggested by the low internalization and overall suppressed cell association values acquired in the presence of excess DG2. 9 Substitution of Glu2–6 by DGlu2–6 had no impact on internalization rates, as shown after comparison of the respective values between [111In]CP04 (Fig. 3A) and [111In-DOTA]MG0 (Fig. 3B) (p > 0.05). On the other hand, truncation of the pentaGlu2–6 repeat in [111In-DOTA]MG11 caused a significant drop (p < 0.001) in internalization rates (Fig. 3C).

Internalization and cell association of [111In]CP04 (,  ; n = 4)
; n = 4)  ,
,  ; n = 3)
; n = 3)  ,
,  ; n = 3)
; n = 3)
Radioligand stability in mice
The in vivo stability of [111In]CP04, [111In-DOTA]MG0, and [111In-DOTA]MG11 was studied in peripheral mouse blood 5 minutes pi using HPLC analysis. Within this period, ≈70% of [111In-DOTA]MG0 and its DGlu2–6 equivalent [111In]CP04 were detected intact in the blood samples (Fig. 4A, C, respectively). In contrast, truncated [111In-DOTA]MG11 was rapidly degraded, with less than 5% detected intact in blood as previously reported (Fig. 3E). 20 Impressively, treatment with PA increased the levels of circulating [111In-DOTA]MG11 to 70% (Fig. 4F), while even the more stable [111In]CP04 and [111In-DOTA]MG0 profited as well by PA coinjection with the amount of parent radiopeptide detected intact raised to ≈85% (Fig. 4B, D, respectively).
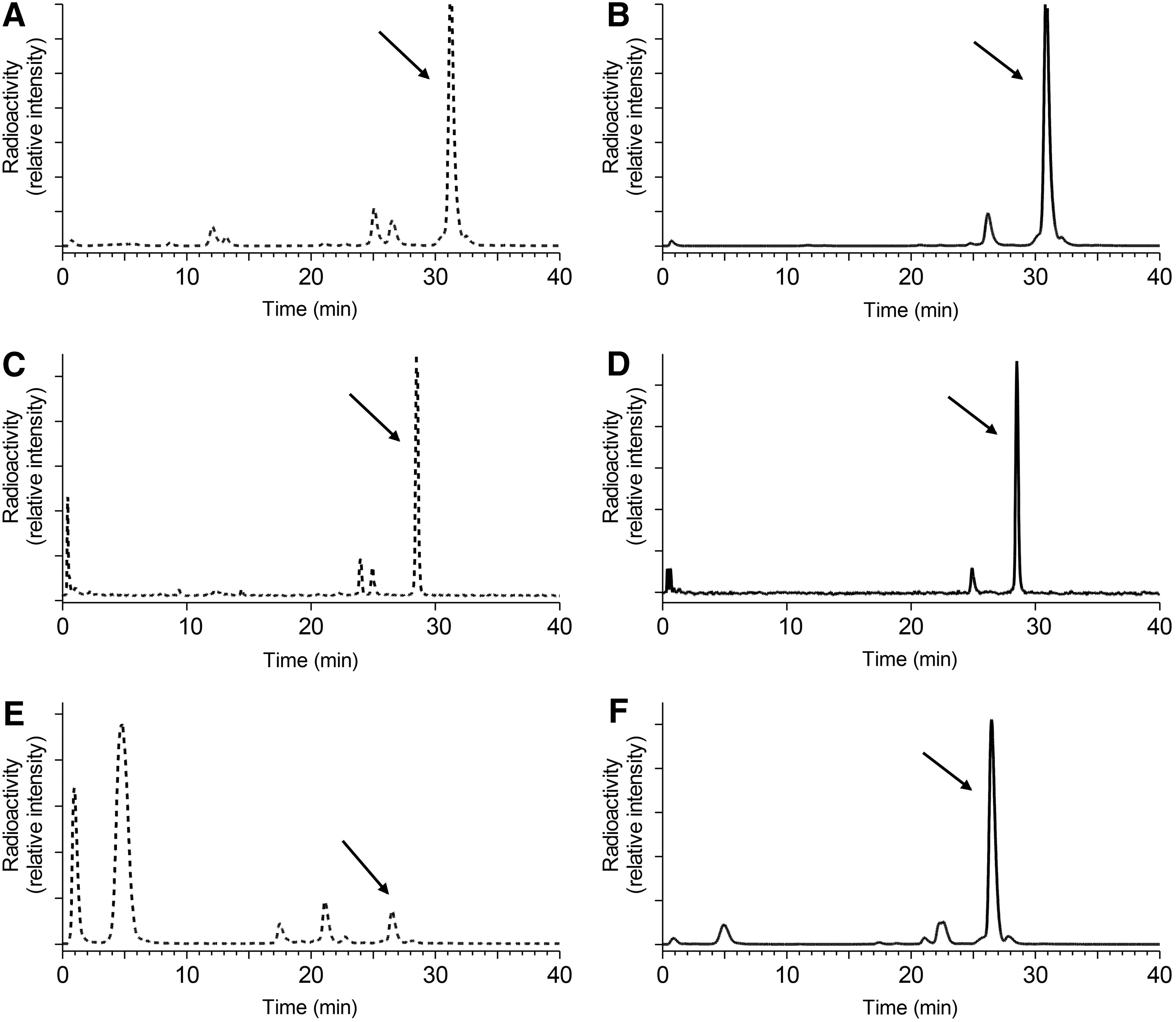
HPLC analysis (system 3) of blood collected 5 minutes pi of [111In]CP04 (∼70% intact)
Radioligand biodistribution in SCID mice bearing A431-CCK2R(+/−) xenografts
The biodistribution of [111In]CP04, [111In-DOTA]MG0, and [111In-DOTA]MG11 was compared in SCID mice bearing a double tumor model: in their right flank, animals carried A431-CCK2R(+) xenografts and, in their left flank, A431-CCK2R(−) tumors devoid of CCK2R expression and thus serving as negative controls. 24 Biodistribution at 4 hours pi involved two separate groups per radioligand, the first comprising mice coinjected with vehicle (control) and the second with PA (PA), as shown in Table 1; data in the table are expressed as mean %ID/g ± SD, n = 5. All three radioligands displayed a rapid clearance from blood and background tissues. While the predominant excretion pathway was through the kidneys and the urinary tract, kidney retention significantly differed across analogs. Specifically, [111In-DOTA]MG0 exhibited by far the highest renal uptake (127.04 ± 21.86%ID/g), while these values notably dropped in its DGlu2–6 counterpart [111In]CP04 (6.52 ± 0.63%ID/g) and were minimal in truncated [111In-DOTA]MG11 (1.30 ± 0.29%ID/g). On the other hand, in line with stability findings, [111In-DOTA]MG0 displayed the highest tumor uptake (11.91 ± 2.21%ID/g), followed by [111In]CP04 (8.49 ± 0.39%ID/g), while truncated [111In-DOTA]MG11 exhibited the lowest tumor uptake (2.49 ± 0.92%ID/g). In all cases, the uptake in the A431-CCK2R(−) tumors remained very low (<0.20%ID/g), suggesting a CCK2R-mediated localization process.
Data are expressed as %ID/g tissue for control and PA groups and values represent mean ± SD, n = 5.
In agreement with results previously reported. 20
Coinjection of PA.
Highly significant (p < 0.001).
Significant (p < 0.01) difference between control and PA animal groups (unpaired two-tailed Student's t-test).
PA, phosphoramidon.
Treatment with PA had a prominent impact in almost all above values, presumably as a direct result of their stabilization in the circulation. As expected, [111In-DOTA]MG11 was the most critically PA-affected radiotracer, showing an impressive increase in the CCK2R(+) tumor uptake (15.05 ± 1.65%ID/g, p < 0.001). At the same time, the uptake of [111In-DOTA]MG11 in the kidneys and the CCK2R(−) tumors remained low and unaffected by PA treatment (p > 0.05), resulting in the most favorable tumor-to-kidney ratios (Fig. 5). Similar levels of uptake were displayed in the CCK2R(+) tumors after PA coinjection also by [111In]CP04 (16.01 ± 2.31%ID/g) and [111In-DOTA]MG0 (17.18 ± 0.93%ID/g). Of particular interest is the effect of PA treatment on the kidney accumulation of these analogs. Interestingly, [111In]CP04 remained unaffected by PA treatment (7.86 ± 1.61%ID/g, p > 0.05), whereas the unfavorably high renal values of [111In-DOTA]MG0 were significantly elevated even further by PA coinjection (194.71 ± 13.24%ID/g; p < 0.001).

Comparative biodistribution data of [111In]CP04 (,  ), [111In-DOTA]MG0 (
), [111In-DOTA]MG0 ( ,
,  ), and [111In-DOTA]MG11 (
), and [111In-DOTA]MG11 ( ,
,  )
32
in SCID mice bearing twin A431-CCK2R(+) and A431-CCK2R(−) human xenografts at 4 hours pi; patterned bars represent control groups (mice coinjected with vehicle only) and solid bars represent PA groups (mice coinjected with PA). Data expressed as %ID/g ± SD (n = 5) are selectively shown for A431-CCK2R(+) tumors
)
32
in SCID mice bearing twin A431-CCK2R(+) and A431-CCK2R(−) human xenografts at 4 hours pi; patterned bars represent control groups (mice coinjected with vehicle only) and solid bars represent PA groups (mice coinjected with PA). Data expressed as %ID/g ± SD (n = 5) are selectively shown for A431-CCK2R(+) tumors
Discussion
Previous studies have indicated a significant role for radiolabeled minigastrin analogs in the management of patients with CCK2R-positive tumors, and especially in the diagnostic imaging and radionuclide therapy of primary and metastatic MTC.
4
–6,8,11,26
Coupling of a suitable metal chelator at the N-terminus of minigastrin and its analogs has allowed labeling with diagnostic radiometals for SPECT and PET imaging, as well as with therapeutic β-emitters.
9,15,16, 27
–31
Studies at the preclinical and the clinical level have shown that the Glu2–6 repeat in the minigastrin chain is not only associated with higher metabolic stability and better localization in CCK2R-positive tumor lesions but also with unfavorable accumulation in the kidneys.
10,12,15,17
On the other hand, truncation of the Glu2–6 chain, while resolving the problem of high renal accumulation, has led to poor metabolic stability and, consequently, to weak tumor targeting.
10,11,15,17
To tackle this problem, modifications of the Glu2–6 part of the molecule have been attempted.
18,19
Thus, by switching the stereochemistry from
We have recently proposed an effective new strategy to increase the in vivo tumor targeting of peptide radioligands through their stabilization in peripheral blood by coinjection of a suitable peptidase inhibitor. 20 More specifically, by coinjection of the NEP inhibitor, PA, together with somatostatin, bombesin, and gastrin radioligands, we were able to induce impressive amplification of tumor uptake in animal models. This strategy has resulted in higher diagnostic sensitivity and recently also in enhanced therapeutic efficacy of peptide radioligands at the preclinical level. 20,32 –34 By adopting this concept in the case of [111In-DOTA]MG11 and other fast biodegradable desGlu2–6 minigastrin radiotracers obtained by Met 11 substitution, we have consistently achieved unprecedented tumor-to-kidney ratios in mice. 32,35
In the present work, we have been interested to compare the efficacy of this approach across the three aforementioned minigastrin radiotracers, [111In-DOTA]MG0, [111In]CP04, and [111In-DOTA]MG11, which differ in the critical Glu2–6 part of the molecule. As expected, assessment of metabolic stability after injection in mice revealed Glu2–6-related differences, with [111In-DOTA]MG0 and its DGlu2–6 congener [111In]CP04 being significantly more robust in vivo compared with their desGlu2–6 counterpart [111In-DOTA]MG11 (Fig. 4). This situation dramatically changed after coinjection of PA, which strengthened the resistance of all three radiotracers to in vivo degradation by NEP. As expected, the most metabolically vulnerable [111In-DOTA]MG11 profited most by PA (>14-fold increase in stability).
Regarding biodistribution (Table 1 and Fig. 5), the radiotracer rank of tumor-targeting efficacy was found to be [111In-DOTA]MG0 > [111In]CP04 >> [111In-DOTA]MG11 in controls, showing that DGlu2–6 substitution only marginally affected tumor uptake. This finding is in line with the combination of in vitro internalization (Fig. 3) and in vivo stability results (Fig. 4). Furthermore, the radiotracers could be ranked according to renal retention as [111In-DOTA]MG0 >> [111In]CP04 > [111In-DOTA]MG11, revealing favorable tumor-to-kidney ratios for [111In]CP04 and especially for [111In-DOTA]MG11. This discrepancy may be assigned to the distinct features of each radiotracer and its individual interaction with the mouse renal system. For example, in previous studies, [111In-DOTA]MG0 demonstrated high kidney uptake and retention due to reabsorption in the proximal tubules, which was associated with the penta-Glu chain. The exact mechanism of the observed renal retention has not been elucidated yet, but the involvement of polyglutamate negative charges has been hypothesized.
14,36,37
Changing the stereochemistry of the implicated multinegative polyglutamate sequence from
It is interesting to compare the impact of in situ NEP inhibition by PA on the above biodistribution profiles of the three radiotracers, especially focusing on tumor and kidney values. Although the tumor uptake of both [111In-DOTA]MG0 and [111In]CP04 visibly increased by PA treatment (up to 2-fold), the corresponding value for [111In-DOTA]MG11 showed a remarkable greater than 6-fold enhancement. Apparently, the extent of in vivo stabilization induced by PA across analogs determined the respective increase of tumor uptake, being maximum for [111In-DOTA]MG11 and less pronounced for the other two more stable analogs (Table 1). Eventually, after treatment with PA, all three radiotracers exhibited comparable tumor values and thus similar tumor targeting efficacy.
On the other hand, the renal uptake unfavorably increased even further by PA treatment in the case of [111In-DOTA]MG0, while remaining at similar to control levels for both the DGlu2–6-subsituted and the desGlu2–6-truncated analogs. It is reasonable to assume that PA by stabilizing the peptides in peripheral blood only increased the amount of intact radiotracer reaching the kidneys, but thereafter each analog underwent different processing and was trapped ([111In-DOTA]MG0) or not ([111In]CP04 and [111In-DOTA]MG11) after glomerular filtration. This hypothesis is further supported by the distinct impact of PA treatment on the kidney uptake of other radiopeptides as well. For example, the kidney uptake of the Lys9-containing [111In-DOTA]SS14 (111In-DOTA-Ala-Gly-c[Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys]-OH) significantly increased after PA coinjection (from 4.2 ± 0.3%ID/g to 33.1 ± 3.7%ID/g at 4 hours pi). 20 On the other hand, the low kidney uptake of [111In]PanSB1 (111In-DOTA-PEG2-DTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2), containing neither positively charged Lys nor negatively charged polyglutamate residues associated with renal retention, remained unaffected by PA coinjection (from 2.7 ± 0.3%ID/g to 3.4 ± 0.5%ID/g at 4 hours pi). 20
As a result of PA treatment, tumor-to-kidney ratios impressively increased from 1.9 ± 0.5 to 9.6 ± 1.0 only in the case of [111In-DOTA]MG11, but remained significantly lower for the other two Glu2–6-modified radiotracers (Fig. 5 C). Accordingly, in vivo NEP inhibition turned out to be a very powerful tool to improve the pharmacokinetic properties of rapidly biodegradable and not kidney-retained [111In-DOTA]MG11, hitherto not considered for clinical application. However, the present study has shown that in situ NEP inhibition may lead to superior radiotracer pharmacokinetic profiles in preclinical models compared with time- and cost-intensive structural modification approaches, 38 as those adopted for the development of [111In]CP04. It would be most interesting to establish if and to what extent [111In-DOTA]MG11 performs better during in situ NEP inhibition in humans compared with [111In]CP04. While [111In]CP04 is currently undergoing evaluation in MTC patients in a multicenter European clinical trial, the authors have been intensively engaged in initiating a proof-of-principle study of the new NEP inhibition concept in man. Toward this goal, [111In-DOTA]MG11 has been selected as a paradigm compound for human application in combination with a suitable NEP inhibitor. For this purpose, inhibitor type, potency, dose, chemical stability, route of administration, and other parameters related to biosafety and efficacy for successful clinical translation are under investigation. 39
Conclusions
The efficacy of molecular imaging and radionuclide therapy of tumors with the aid of peptide radioligands largely relies on their stability in the biological milieu, especially for the short period of time needed to reach their tumor-associated targets after entering the circulation. The development of in vivo robust radiotracers derived from native peptide motifs is a lengthy and resource-intensive process given that the attempted structural modifications may compromise important properties of end compounds other than metabolic stability, such as receptor affinity and in vivo pharmacokinetics.
Based on the prominent role of NEP in initiating the in vivo degradation of many peptide radioligands of interest to nuclear oncology, the authors have proposed the coinjection of a NEP inhibitor to improve radiopeptide bioavailability, in vivo tumor targeting, and overall pharmacokinetics. This approach, has turned out to be particularly successful in the case of [111In-DOTA]MG11 and undeniably warrants further validation in humans. A first successful application of biodegradable [111In-DOTA]MG11 in humans after in situ stabilization by a NEP inhibitor will serve as a paradigm to establish this simple approach in the clinic and will highlight its strength of bypassing the need for extensive and costly development of compound libraries.
Footnotes
Acknowledgement
Partial financial support has been provided by the ERA-NET program GRAN-T-MTC.
Disclosure Statement
No competing financial interests exist.