
Abstract
Hepatitis B X-interacting protein (HBXIP) has been found overexpressed in several types of human cancer, however, the status of HBXIP expression in urothelial carcinoma of the bladder (UCB) has not been explored. In this study, the authors used real-time polymerase chain reaction and Western blot to test the expression of HBXIP in UCB and adjacent tissues. The expression of HBXIP was significantly increased in UCB tissues. In addition, they showed that suppression of HBXIP induced cell cycle arrest and increased cell apoptosis in T24 cells. Also, suppression of HBXIP also decreased T24 and PC3 cell proliferation, migration, and invasion. More importantly, the authors found that inhibition of HBXIP reduced the tumorigenesis in vivo, suggesting that HBXIP plays an important role in UCB progression. These data for the first time showed that HBXIP acts as an oncoprotein in UCB, suggesting that HBXIP may become a potential novel therapeutic target for the treatment of UCB.
Introduction
Urothelial carcinoma of the bladder (UCB) is the most common histopathological type and accounts for ∼90%–95% of all bladder tumors. 1,2 The incidence of bladder tumors has become secondary to an increase in smoking in Asia. 3 In China, bladder cancer is the fifth most common cancer in men and the most common malignancy in the genitourinary tract. 4 Approximately 50% of UCB patients develop metastatic/recurrent disease within 2 years. 5 Although a number of molecular markers with possible prognostic significance have been discussed in the literature, 6 the effective molecular markers for the treatment of UCB are limited. Therefore, a specific molecular target for UCB treatment needs to be clarified.
Hepatitis B virus is a small DNA (about 3.4 kb) virus containing four partially overlapping open reading frames, encoding the C, S, and X proteins and a viral DNA polymerase. 7 Among these four proteins, only the X protein (known as HBx) is clearly associated with tumorigenesis. 8 Mammalian hepatitis B X-interacting protein (HBXIP) is a conserved protein of unknown function, which is originally identified because of its interaction with the C terminus of HBx. 9 Previous study revealed that HBXIP plays a critical role at specific steps involved in mitosis and cell division, particularly in centrosome duplication to form a bipolar spindle during prometaphase and at telophase for cell splitting to form two daughter cells. 10 Recently, accumulating evidences show that HBXIP acts as an oncoprotein in controlling cell proliferation, apoptosis, and division. In addition, HBXIP imports into the nucleus of breast cancer cells, acting as a transcriptional coactivator to promote the progression of breast cancer. 11 –16 Moreover, HBXIP as a coactivator is involved in the transcription of MDM2 mediated by p53, and HBXIP enhances MDM2-mediated p53 degradation, leading to the growth of breast cancer. 17 Furthermore, HBXIP has been found as a target of miR-520, involved in the development of hepatocarcinogenesis. 18 More recently, HBXIP has been reported to stimulate the migration of ovarian cancer cells and breast cancer cells by activating transcriptional factor Sp1. 19,20
In this study, they found that the expression of HBXIP was significantly increased in UCB. Suppression of HBXIP by siRNA inhibited cell proliferation, induced cell cycle arrest, and increased cell apoptosis. In addition, inhibiting the expression of HBXIP reduced cell migration and invasion. More importantly, knocking down HBXIP decreased the tumorigenesis in vivo. These data for the first time showed that HBXIP acts as an oncoprotein in UCB, suggesting that HBXIP may become a potential novel therapeutic target for the treatment of UCB.
Materials and Methods
Clinical specimens
Pertinent patient clinical reports were obtained with patient consent and the approval of the institutional Clinical Ethics Review Board. All patients have signed a written informed consent form. All of 20 cases of primary UCB and their adjacent tissues and 20 cases of primary prostate cancer tissues and their adjacent tissues were collected from the time of original diagnosis and kept in liquid nitrogen for RNA extraction.
Cell culture
UCB cell lines, including SV-HUV-1, RT4, 253J, and T24, prostate cancer cell line PC3 and 293T cells were obtained from American Type Culture Collection (ATCC) and cultured in RPMI-1640 supplemented with 10% fetal bovine serum (Hyclone) with appropriate antibiotics at 37°C in 5% CO2 humidified incubator.
Reverse transcription–polymerase chain reaction
Total RNA was extracted by using Trizol reagent (TAKARA). The first-strand cDNA was synthesized from total RNA by using a reverse transcription kit (BestarqPCR RT kit; DBI). The primer sequences for HBXIP were 5′ GAGCCCAAGCCTTCGTCAG 3′ and 5′ GGCACGTCCTTCTCCACCA 3′, and the primer sequences for Actin were 5′ ATCGTGCGTGACATTAAGGAGAAG 3′ and 5′ AGGAAGGAAGGCTGGAAGAGTG 3′. A total volume of 25 μL quantitative real-time polymerase chain reaction (qPCR) mixture contained 10 μL of 2× SYBR green PCR mix (DBI), 0.2 μM of primers, and 1 μL of cDNA template. Cycling parameters were as follows: initial denaturation at 94°C for 2 minutes, followed by 40 cycles of denaturation at 94°C for 20 seconds, annealing at 58°C for 20 seconds, and extension at 72°C for 20 seconds. Melting curve analysis was performed at temperature ranging from 62°C to 95°C with stepwise fluorescence acquisition at every 1°C/s. All samples were repeated thrice. The expression of HBXIP was calculated by relative quantification using Actin as the endogenous reference gene.
Western blot analysis
Primary tissues and cells were lysed on ice in the RIPA lysis buffer. BCA method was used to determine the protein concentration. Equal amount of tissue or cell extracts was subjected to electrophoresis in SDS-PAGE and transferred to PVDF membrane for antibody blotting. Mouse anti-β actin was from Ambion and Rabbit anti-HBXIP was from Abcam.
siRNA transfection
Cells were seeded onto six-well plates. After 16 hours, cells were transfected with HBXIP and control siRNA. In each well, 100 nM of HBXIP (5′-GUGUCUAGAAUCAGAUAAUdTdT-3′) or negative control (5′-UUCUCCGAACGUGUCACGUdTdT-3′) siRNA and 10 μL of Lipofectamine 2000 (Invitrogen) were added to Opti-MEM (Life Technologies) and mixed. After 10–15 minutes of incubation, the mixture was gently added to the cells. The plate was incubated for an indicated time until further assay.
Flow cytometry analysis
Cells were seeded onto six-well plates and transfected with control or HBXIP siRNA. After 48 hours, cells were collected and suspended in ice-cold 70% ethanol for 30 minutes (fixation) and labeled with propidium iodide (PI, 50 μg/mL; Sigma) for at least 20 minutes in dark. The cell cycle distribution was analyzed directly on a Becton Dickinson FACScan.
Annexin V assay
Cells were transfected with control or HBXIP siRNA. After 48 hours, cells were collected. Annexin V and propidium iodide (Annexin V-FITC apoptosis Detection Kit; Merck) were added to each sample according to manufacturer's protocol. Cells were analyzed by flow cytometry within 1 hour.
Cell proliferation assay
Total of 5000–10,000 cells were seeded onto 96-well flat-bottom plates and transfected with control siRNA or HBXIP siRNA. After 24, 48, or 72 hours, 10 μL of CCK-8 solution was added to each well of the plate and incubated for 1–4 hours. The absorbance at 450 nm was measured using a microplate reader.
Transwell migration and invasion assay
Cells were starved overnight in an assay medium (RPMI-1640 without FBS). Top chambers of 24-well plates were pretreated with 1% Matrigel (for migration assay; BD) or 10% Matrigel (for invasion assay) in PBS and incubated for 1 hour at room temperature. After incubation, top cells were removed and bottom cells were fixed and stained with crystal violet to visualize nuclei. The number of migration or invasion cells in three to five fields was counted.
Stable cell line establishment
T24 cells stably suppressing HBXIP or control were constructed with a lentivirus vector (pLKO.1-puro). In brief, shNC or shHBXIP was subcloned to pLKO.1-puro and cotransfected with psPAX2 and pMD2.G to 293T cells for virus production. T24 cells were infected by shControl or shHBXIP lentivirus and selected by puromycin (2 μg/mL).
Mouse xenograft assay
Six-week-old BALB/c athymic nude mice were used for the xenograft assay. Mice were subcutaneously injected with shHBXIP T24 cells (5 × 106 cells per mouse). The tumor volume was measured every 7 days. Volumes were estimated using the formula V = (π/6) × ab2, where a is the longer of the two dimensions and b is the shorter one. After 35 days, mice were sacrificed and the xenografts were collected.
Statistics
Statistical analyses were done using SPSS v. 19.0. Unpaired t-tests or Mann–Whitney U tests were used to compare the two groups. All p-values quoted are two sided. p < 0.05 is considered statistically significant.
Results
HBXIP is overexpressed in UCB
The authors obtained 20 cases of primary UCB and their adjacent tissues and used real-time PCR to investigate HBXIP expression in UCB and their adjacent tissues. Results showed that HBXIP was significantly increased in UCB compared with their adjacent tissues (p < 0.001, Fig. 1A). Next, the authors measured the expression of HBXIP in four UCB cancer cell lines, SV-HUC-1, RT4, 253J, and T24 by using real-time PCR and Western blotting. They found that in T24 cells, the mRNA and protein expression of HBXIP was the highest among these four cell lines (Fig. 1B, C).

HBXIP is overexpressed in UCB.
Suppression of HBXIP by siRNA in T24 cells
The authors genetically suppressed endogenous HBXIP by different doses of siRNA. HBXIP mRNA expression was substantially reduced at 48 hours in a dose-dependent manner (Fig. 2A). The mRNA expression of HBXIP was inhibited significantly (>70%) at 100 nM of siRNA (p < 0.001, Fig. 2A). They also detected the protein expression of HBXIP and found that the protein level of HBXIP was significantly decreased at 72 hours after transfection with 100 nM siRNA (Fig. 2B).
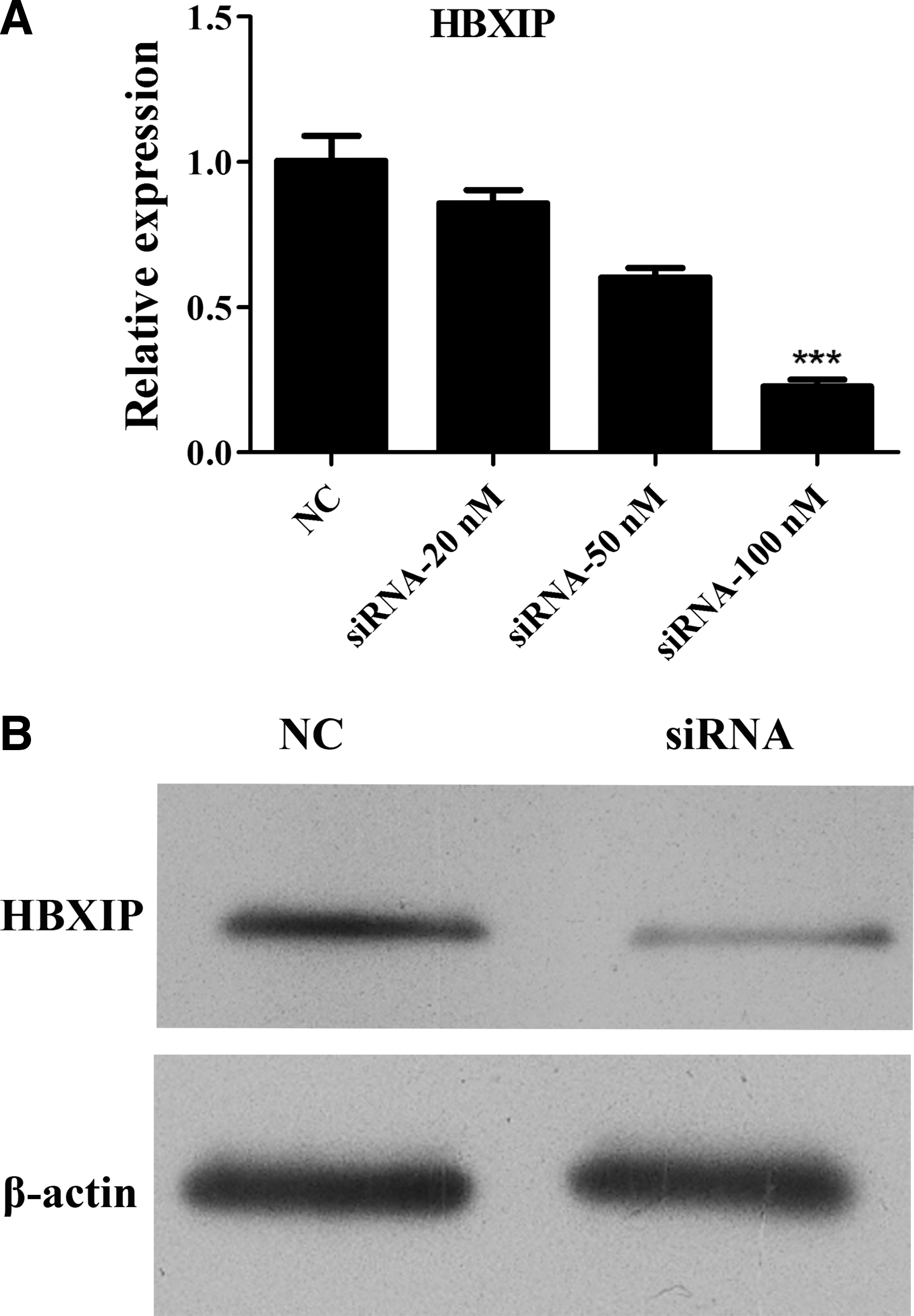
Suppression of HBXIP by siRNA in T24 cells.
Inhibition of HBXIP induces cell apoptosis and cell cycle arrest in UCB cells
Next, the authors detected the cell apoptosis in HBXIP siRNA or control siRNA-treated T24 cells. Annexin V assay revealed that HBXIP siRNA induced both early and late apoptosis significantly compared with control siRNA (p < 0.01, Fig. 3A). In addition, they measured the cell cycle distribution by flow cytometry. T24 cells were transfected with HBXIP siRNA or control siRNA; after 48 hours, cells were collected for cell cycle analysis. As shown in Figure 3B, HBXIP siRNA significantly induced cell cycle arrest at G1 phase (p < 0.01). However, cells at G2/M and S phases were decreased in HBXIP siRNA-transfected cells (p < 0.05). The results suggested that HBXIP induced cell apoptosis by G1 cell cycle arrest.

Inhibition of HBXIP induces cell apoptosis and cell cycle arrest in T24 cells. T24 cells were seeded onto six-well plates and transfected with control or HBXIP siRNA. After 48 hours, cells were collected for
HBXIP siRNA reduces cell proliferation, migration, and invasion in vitro and tumorigenesis in vivo
The authors addressed if inhibition of HBXIP expression by siRNA would lead to the reduction of cell proliferation. T24 cells were transfected with HBXIP siRNA or control siRNA. As shown in Figure 4A, inhibition of HBXIP by siRNA suppressed growth rate of T24 cells in a time-dependent manner by the CCK8 assay. HBXIP siRNA decreased cell proliferation to nearly 50% after 72 hours (p < 0.01). Similarly, data showed that HBXIP siRNA significantly reduced the cell proliferation in prostate cancer cell line PC3. They also asked if inhibition of HBXIP by siRNA would attenuate T24 cell migration and invasion. Transwell migration assay revealed that HBXIP siRNA potently inhibited >40% migration capacity of T24 cells (p < 0.01, Fig. 4B, upper panel). In addition, the transwell invasion assay showed that HBXIP siRNA effectively prevented >50% of cells (p < 0.01, Fig. 4B, lower panel) from crossing the Matrigel-coated transwell membrane pore, suggesting an important role of HBXIP in cancer cell proliferation, migration, and invasion. Moreover, the migration and invasion capacity of PC3 cells were also significantly inhibited by HBXIP siRNA. Furthermore, the authors tested the effect of HBXIP inhibition on tumorigenicity in vivo by using a xenograft tumor model. As shown in Figure 4C, shHBXIP T24 cells significantly reduced tumor growth compared to control T24 cells, the data suggesting that inhibition of HBXIP reduced the tumorigenicity of UCB.
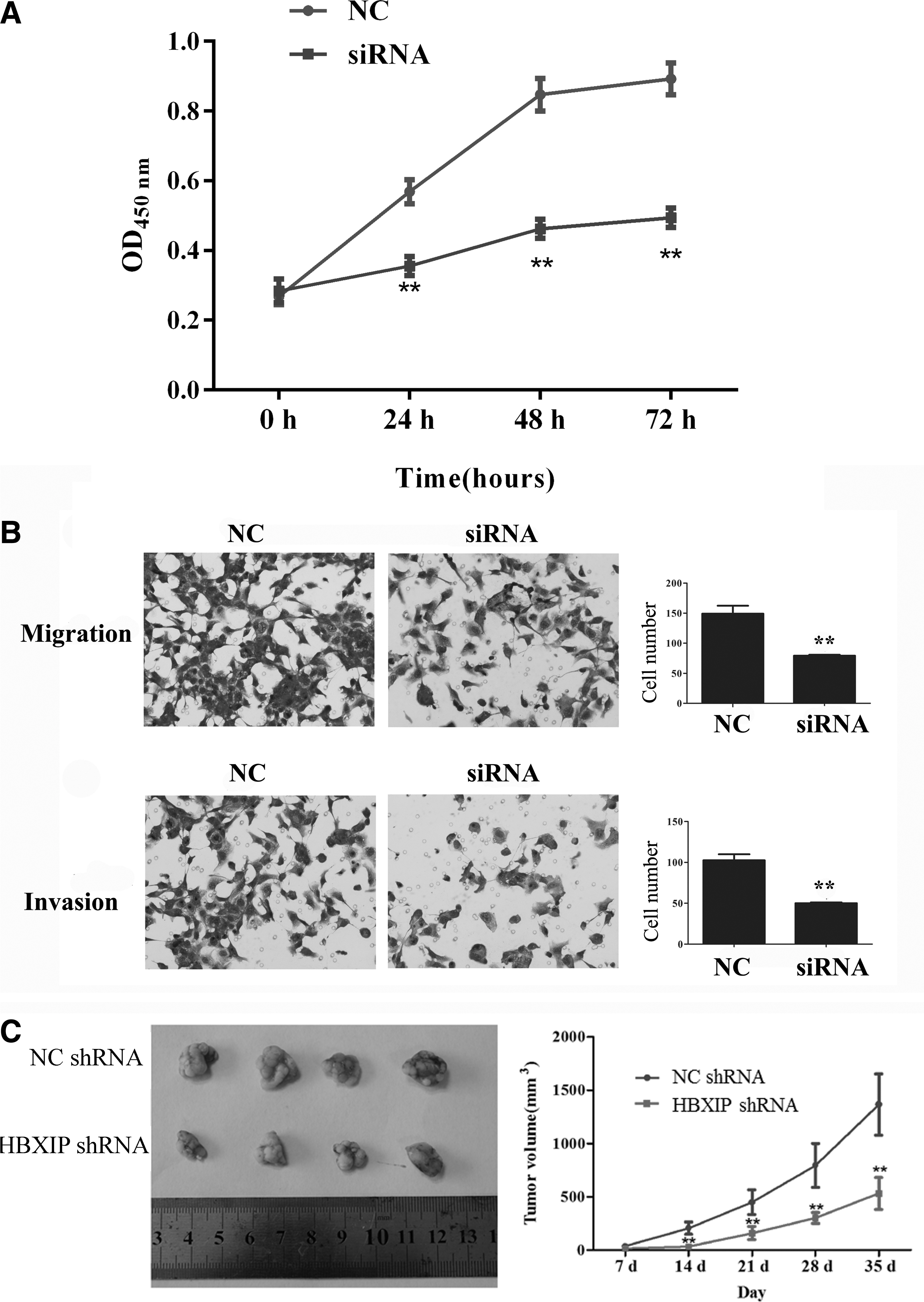
HBXIP siRNA reduces cell proliferation, migration, and invasion in vitro and tumorigenesis in vivo.
Discussion
HBXIP plays a critical role in promoting the proliferation and migration of breast cancer cells by acting as a coactivator of transcription factors such as E2F1, TFIID, CREB, and Sp1. 11 –16 However, HBXIP in UCB has not been studied. In this study, the authors reported that HBXIP was upregulated in UCB tissues compared to adjacent normal tissues. In addition, they showed that specific inhibition of HBXIP by siRNA suppressed UCB cell growth and induced apoptotic cell death. Most importantly, the authors found that suppression of HBXIP potently reduced cell migration, invasion, and tumorigenesis. These data have provided clear evidence that HBXIP plays important roles in cell proliferation and migration in UCB cells. Collectively, this study suggested that HBXIP may serve as a potential molecular target for the treatment of UCB.
UCB, especially the nonmuscle invasive type, is known to be very expensive to treat and follow. 21 Therefore, searching for a new therapeutic target for effective treatment approaches will offer great promise in improving the current treatment. In this study, the authors provided evidence that HBXIP was overexpressed in UCB tissues (Fig. 1). The results were consistent with previous reports, which showed that the expression of HBXIP increased in breast cancer tissues, 14,16,17 hepatocellular carcinoma tissues, 18 and ovarian cancer tissues. 19 Data suggested that HBXIP may serve as a prognosis marker of UCB.
To determine the biological relevance of HBXIP in UCB, they performed functional assays. Suppression of HBXIP resulted in significant inhibition of cell proliferation and induction of cell apoptosis and cell cycle arrest (Figs. 3 and 4). HBXIP has been found as an interaction partner of Survivin, a BIR (baculoviral IAP repeat) family chromosomal passenger protein involved in controlling apoptosis and cell division, 22 and collaborates with cytosolic Survivin to suppress apoptosis in interphase cells. 23 In addition, HBXIP upregulates Lin28B to promote proliferation of breast cancer cells. 14 Moreover, HBXIP is involved in angiogenesis of hepatoma HepG2 cells 24 and enhances angiogenesis and growth of breast cancer through modulating fibroblast growth factor 8 (FGF8) and vascular endothelial growth factor (VEGF). 16 A recent report shows that highly expressed HBXIP accelerates the MDM2-mediated degradation of p53 in breast cancer by modulating the feedback loop of MDM2/p53. 17 In this study, the authors for the first time showed that HBXIP played a role in UCB cell proliferation. The mechanism of HBXIP in promoting UCB cell growth needed to be identified further.
Two-thirds of noninvasive bladder cancers will eventually have a recurrence within 3 years of the initial diagnosis with a 25% risk of progression to muscle-invasive tumors requiring aggressive treatment. 25,26 In this study, the authors found that suppression of HBXIP significantly decreased UCB cell migration, invasion, and tumorigenesis (Fig. 4), suggesting that HBXIP may serve as a metastatic target for the therapy of UCB. Previously, several studies showed that HBXIP promoted breast cancer cell migration by various mechanisms, such as upregulating s100a4, 11 general control non-derepressible 5 (GCN5)-mediated microtubule acetylation, 27 increasing filopodia formation, 28 and activating transcriptional factor Sp1. 20 The authors reported in this study that HBXIP contributed to UCB cell migration and invasion. Although they did not clarify the mechanism of HBXIP in UCB cell migration and invasion, the data presented that HBXIP may be served as a metastatic target for UCB treatment.
In conclusion, these data for the first time identified that inhibition of HBXIP contributed to the tumor suppression by reducing cell proliferation, migration, invasion, and tumorigenesis. HBXIP might be developed as a therapeutic target in the urothelial carcinoma of the bladder.
Footnotes
Acknowledgment
This work was supported by YanBian University Science and Technology Department Item (201410).
Disclosure Statement
There are no existing financial conflicts.