
Abstract
Autophagy is a devouring process during which cytoplasmic proteins, organelles, or other contents are phagocytized and delivered into an autophagosome, which then fuses with a lysosome to become an autolysosome. Autophagy is involved in various human diseases, including cancers, and is triggered and regulated by different signaling pathways under various stimuli such as oxidative stress. Oxidative stress is caused by excessive reactive oxygen species (ROS). It is well known that mitochondria are the primary cellular sources of ROS, which play important roles in the induction of cell death after radiation treatment. It is unknown whether ROS participate in autophagy regulation, and the underlying mechanism is still unclear. In this review, the authors focus on the association between mitochondrial stress and autophagic cell death. They also discuss the roles of ROS and the lysosomal–mitochondrial axis.
Introduction
Autophagy is an evolutionary cellular process during which cytoplasmic proteins or organelles are degraded and recycled. 1 –3 Under various cellular stresses, autophagy will lead to either adaptive response or cell death. 4 On one hand, appropriate autophagy facilitates tumor progression due to its basic function in maintaining cell metabolism. On the other hand, massive autophagy may lead to type II programmed cell death (PCD). 5
Mitochondria usually act as cellular power houses. Cellular energy, ATP, is produced via a specialized respiratory oxidative phosphorylation system (OXPHOS), which consists of five complexes (I–V) in the mitochondrial inner membrane. A functional mitochondrial OXPHOS plays vital roles in cell survival. However, mitochondria are also the major intracellular sources of reactive oxygen species (ROS). Under diverse external stimuli, such as oxidative stress, starvation, and ionizing radiation (IR), 6 ROS production will increase. 7 In mammalian cells, mitochondrial ROS molecules participate in pathway regulation and even in determining the fate of cells. 8
It is known that some mitochondrial proteins, such as cytochrome c and apoptosis inducing factor (AIF), can be released from mitochondria to trigger apoptosis. As such, mitochondria play key roles in activating apoptosis in mammalian cells. A number of recent findings have verified that the specific function of mitochondria has differential effects on autophagy. However, the roles of mitochondria in autophagy regulation still need to be clarified. In this review, the authors attempt to elucidate the association between mitochondria, ROS generation, and autophagic cell death.
Function of mitochondria
Mitochondria have a double-membrane architecture, consisting of an outer mitochondrial membrane and an inner mitochondrial membrane (IMM). The IMM is organized in characteristic folds that protrude into the mitochondrial matrix and accommodate the respiratory chain complexes. Mitochondria are the primary organelles for energy-generating metabolism in eukaryotic cells and are critical for numerous metabolic pathways. 9 Three stages are involved in the energy-generating process. The first stage, known as glycolysis, takes place in the cytoplasm and generates only a small amount of energy. The second stage, called the citric acid cycle, takes place in the mitochondrial matrix and also generates a small amount of energy. The last stage, named oxidative phosphorylation, occurs in the mitochondrial inner membrane and generates 90% of the cellular energy via an electron transport chain (ETC) and ATPas. 10
Mitochondrial status strongly effects not only energy production but also a number of intracellular signals. Mitochondrial Ca2+ transport has significant effects on cytoplasmic Ca2+ signals and cell death pathways. 11,12 Mitochondrial calcium uptake1 (MICU1), identified as an IMM protein, is required to preserve normal mitochondrial calcium levels (Ca2+ m) under basic conditions. 13 Knockdown of MICU1 leads to the accumulation of Ca2+ m, excessive mitochondrial ROS generation, and apoptotic cell death. In addition to Ca2+ signaling, redox reactions can also be regulated by mitochondria. For example, H2O2 and O2 ·-are generated by the ETC within the IMM, while at the same time various antioxidant enzymes within mitochondria such as glutathione peroxidase (GSH-Px), catalase, and superoxide dismutase (SOD) convert ROS to less harmful substances. The balance of redox reactions is important for cell survival, 14 as an excessive ROS level challenges the ability of mitochondria to maintain mitochondrial function and cell viability.
The connections between mitochondria and apoptosis have been explored for many years. Mitochondria contain a series of apoptosis-associated proteins such as cytochrome c, AIF, and Smac/DIABLO. Under certain stresses such as hypoxia, 15 these proteins are released from the mitochondrial intermembrane and enter the cytoplasm. Zheng et al. demonstrated that the flavonoid uvangoletin induced apoptosis in HL-60 cells by promoting the transference of cytochrome c from mitochondria to the cytoplasm. 16 In addition, some studies suggested that release of cytochrome c can be inhibited by Bcl-2. 17,18 However, in the Xenopus system, changes in the mitochondrial membrane potential (ΔΨm) have nothing to do with the release of cytochrome c and its inhibition by Bcl-2. 17 –19 Bcl-2 can impede the release of AIF from mitochondria by preventing mitochondrial permeability transition, these processes are caspase-independent. 20,21
Autophagy, mitophagy, and cell death
Autophagy
Autophagy, also known as “self-digestion,” is a devouring process for recycling basic metabolic units, including amino acids. 22,23 Autophagy can be triggered by a series of events, including nutrient deprivation and exposure to oxidative stress. There are three types of autophagy: microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy. Microautophagy involves direct lysosomal engulfment of cytoplasmic constituents, which are subsequently degraded within the lysosomal lumen. CMA involves the delivery of proteins directly to the lysosome via chaperones such as Hsc70, which can unfold proteins with consensus KFERQ sequences. Hsc70 also recognizes the lysosomal-associated membrane protein 2 (LAMP2A, a lysosomal membrane receptor), which mediates protein translocation into the lysosomal lumen. 24 Macroautophagy, the representative type of autophagy, is a nonspecific degradation process that involves the formation of double-membrane vesicles called autophagosomes and separates the enclosed components from the rest of the cytoplasm. Ultimately, autophagosomes acidic hydrolases (degrade the enclosed components, especially the proteins).
Currently, two kinds of complexes make a critical difference in autophagy regulation. These are the mammalian target of rapamycin (mTOR) complex and the Beclin-1 complex (Fig. 1). Primarily, mTOR acts as a serine/threonine protein kinase and plays a central role in regulating cell growth, survival, division, and motility. 25 The activation of mTOR has an intimate connection with autophagosome formation and maturation. mTORC1 and mTORC2 are two classes of mTOR signaling complexes. Once activated, mTORC1 can negatively regulate the initiation of autophagy via the ULK1 complex, which contains Atg13, Atg101, and FIP200 in mammalian cells. 26 Lamming et al. have demonstrated that mTORC2 facilitates cell viability through Akt. 27 As part of the mTOR pathway, phosphatidylinositol −3-kinase class I (Class I PI3K) is an important player protein in autophagy regulation. When Class I PI3K is activated, Akt (also known as protein kinase B) and phosphoinositide-dependent protein kinase 1(PDK1) tend to be recruited to the cell membrane. Subsequently, mTOR serves as the downstream effective molecular of Akt once it has been phosphorylated by PDK1. 28 In addition, previous research has found that suppression of Akt/mTOR/p70 pathways participates in IR-induced autophagy, which is regulated by HIF-1α. 29 Furthermore, in the absence of growth factor or nutrition, adenosine monophosphate-activated protein kinase (AMPK), an upstream effector of mTOR, can be activated by AMP and positively regulate autophagy by activating the tuberous sclerosis complex (TSC) or directly activating the ULK1complex. 30 –32 It has since been demonstrated that the TSC, negatively regulated by AKT, has an opposite effect on mTOR activation. In prostate cancer, overexpression of Rheb cooperates with PTEN and promotes tumorigenesis. 33 TSC deficiency can increase Rheb activation, which can inhibit autophagy. 34
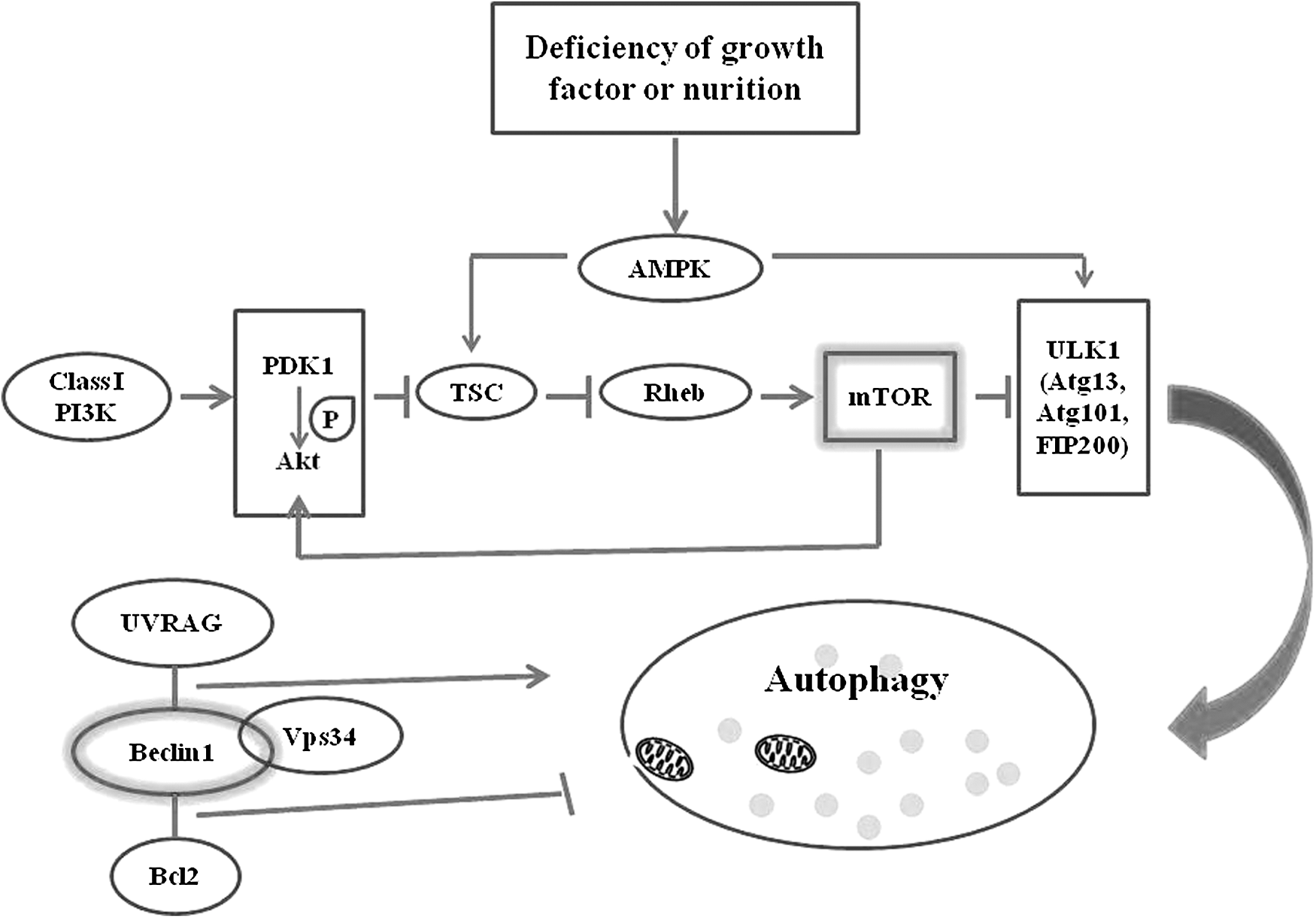
mTOR complex and Beclin 1 complex participate in autophagy. When Class I PI3K is activated, Akt and PDK1 tend to be recruited to the cell membrane. mTOR serves as the downstream effector molecule of Akt, which is phosphorylated by PDK1. The TSC-Rheb pathway, negatively regulated by AKT, is crucial for mTORC1 activation. TSC deficiency improves Rheb activation, which can inhibit autophagy. mTORC1 and mTORC2 are two different complexes of mTOR. Once activated, mTORC1 can negatively regulate the initiation of autophagy via the ULK1/2 complex, which contains Atg13, Atg101, and FIP200 in mammalian cells. The Beclin 1 complex consists of Bcl-2, Beclin-1, UVRAG, and Vps34. Bcl-2 can interact with the Beclin-1 BH3 domain to suppress autophagy; however, UVRAG interaction with the Beclin-1 CCD domain facilitates autophagy initiation. What is more, UVRAG can also enhance the interaction of Beclin-1 and Vps34 and consequently promote the occurrence of autophagy. CCD, central coiled-coil domain; mTOR, mammalian target of rapamycin; TSC, tuberous sclerosis complex; UVRAG, UV radiation resistance-associated gene protein.
Beclin-1 is a 60 kDa coiled-coil protein that contains three domains: a Bcl-2-homology-3 (BH3) domain, a central coiled-coil domain (CCD), and an evolutionarily conserved domain. Beclin-1 is the first mammalian autophagy protein to be described and plays an important role in the initiation of autophagy. Bcl-2 can interact with Beclin-1's BH3 domain to suppress autophagy, although Bcl-2 is always considered as an antiapoptosis protein. 35 UV radiation resistance-associated gene protein (UVRAG) is a tumor suppressor that is frequently mutated in human colon cancer cells. Interaction of UVRAG with Beclin-1's CCD domain facilitates the process of autophagy, and consequently, the proliferation and tumorigenicity of human colon cancer cells are suppressed. 36 Study has suggested that Bif (also known as endophilin B1) can interact with the UVRAG–Beclin-1 complex and serves as an activator of autophagy. 37 Beclin-1 performs as part of a core complex that contains vacuolar sorting protein 34 (VPS34), a class III phosphatidylinositol-3 kinase. UVRAG can also enhance the interaction of Beclin-1 and Vps34, and consequently promotes the initiation of autophagy. 38 A recent study has shown that both Cdk1 and Cdk5 can reduce Vps34 activity through phosphorylation, which thereby inhibits the formation of autophagosomes. 39
In addition to Beclin-1, other signals, including autophagy-related (Atg) proteins and the p53 gene, also take part in the induction of autophagy. Atg proteins play a major role during different stages of autophagy, such as the formation, expansion, and fusion of autophagosomes. 40,41 To date, 38 members of the Atg family have been reported to be primarily required for autophagy. 42 For instance, the class III PI3 kinase complex, including Beclin-1/Atg6, is required for the generation of preautophagosome structures. 43 Other Atg proteins, such as the Atg5-Atg12 protein complex, are also essential for the formation of the autophagosome membrane. When cells are treated with sedanolide, nuclear p53 and NF-κB signaling pathways are upregulated and then autophagy occurs. 44 Vacuole membrane protein 1 (VMP1) is highly activated in acute pancreatitis, and VMP1 expression triggers the occurrence of autophagy and recruits microtubule-associated protein 1 light-chain 3 (MAPLC3). 45 These changes are suppressed by 3-methyladenine (3-MA), an autophagy inhibitor. Furthermore, nutrient or growth deprivation, endoplasmic reticulum (ER) stress, and oxidative stress all potently induce autophagy.
Mitophagy
In the above account, mitochondria are powerful organelles that act as platforms for many crucial processes such as energy conversion and calcium homeostasis. 46 In contrast, mitochondria are also highly vulnerable organelles in response to damage stimuli such as starvation and oxidative stress. Once damaged, mitochondria concomitantly generate massive ROS as by-products, which can enhance mitochondrial dysfunction. Therefore, it is critical for cell homeostasis to eliminate damaged mitochondria. This tends to occur via engulfment and digestion by autolysosomes in a process known as mitophagy. 47 Numerous studies have demonstrated that mitophagy contributes to mitochondrial quality and quantity control. In addition, mitophagy is a highly regulated process and requires the coordinated functions of mitochondrial and cytosolic proteins. 48 In this section, some specific genes, which might participate in the regulation of mitophagy, are listed (Fig. 2).

Mitophagy mechanism. PINK1 is a ubiquitin kinase located in mitochondrial membranes. When mitochondrial damage occurs, PINK1 recruits the E3 ubiquitin ligase parkin to mitochondria and then phosphorylates parkin, consequently initiating mitophagy. Parkin is able to ubiquitinate multiple mitochondrial substrates such as Mitofusin (Mfn1/2), VDAC, and TOM proteins. P62 participates in the PINK1/Parkin pathway by binding to ubiquitinated mitochondrial proteins and stimulating mitophagic degradation. In addition, the AMPK-ULK1, MAPK1/MAPK14 and FUNDC1 signaling pathways were involved in mitophagy. AMPK, adenosine monophosphate-activated protein kinase; VADC, voltage-dependent anion-selective channel; TOM, translocate of the outer membrane.
PTEN-induced putative kinase 1(PINK1) is a serine/threonine ubiquitin kinase 49 located in mitochondrial membranes. PINK1 is usually unstable due to presenilin-associated rhomboid-like protease activity. When mitochondrial damage occurs, PINK1 recruits the E3 ubiquitin ligase parkin to mitochondria and then phosphorylates parkin, consequently initiating mitophagy. 50 In this process, parkin has been shown to be able to ubiquitinate multiple mitochondrial substrates such as mitofusin (Mfn1/2), 50 mitochondrial outer membrane protein, as voltage-dependent anion-selective channel protein 1(VDAC), 51 and the translocate of the outer membrane, leading to mitophagy. Simultaneously, P62 participates in the PINK1/Parkin pathway and is recruited to mitochondria by binding to the ubiquitinated proteins, and finally, the mitochondria are destined for mitophagic degradation. 52 –54 A recent report has shown that mitophagy is regulated by mitochondrial deubiquitinating enzymes, USP30, via the delay of PARK2 (a gene name that encodes Parkin E3 ligase protein) recruitment to mitochondria during mitophagy. 55 Clec16a can interact with Nrdp1 and restrain mitophagy through the Nrdp1/Parkin pathway. 56 As is known, unc-51-like autophagy activating kinase 1 (ULK1) takes part in the initiation of mitophagy. The phosphorylation of ULK1 is regulated by AMPK, which is activated by thyroid hormone (T3)-induced ROS. 32
Protein deglycase DJ-1, also known as Parkinson disease protein 7, is a protein that in humans is encoded by the PARK7 gene. DJ-1 plays a vital role in maintaining mitochondrial membrane potential and preserving mitochondria from fragmentation. In DJ-1 knockdown cells, the accumulation of MAPLC3 punctate around damaged mitochondria obviously increases, 57 which is independent of PINK1, suggesting that both PINK1and DJ-1 can regulate mitochondrial function and autophagy in parallel.
In addition, Bcl-2 family members Nix (also called BNIP3) 58 and BNIP3 are involved in mitochondrial clearance during starvation and play roles in transferring damaged mitochondria into autophagosomes by directly interacting with MAPLC3 or gamma-amino butyric acid receptor-associated protein in mammalian cells. Besides, BNIP3 can interact with MAPLC3 and activate mitophagy. 59 Recently, studies have shown that the MAPK1 and MAPK14 signaling pathways are involved in mitophagy through an alternative autophagy pathway in mammalian cells. 60 In addition, FUN14 domain containing 1 (FUNDC1), an integral mitochondrial outer-membrane protein, is a receptor for hypoxia-induced mitophagy. In one study, FUNDC1 downregulation significantly inhibited hypoxia-induced mitophagy, which was reversed by wild-type FUNDC1. 61
Autophagic cell death
Cell death, either active or passive, occurs in both normal and cancer cells. Passive death is a pathological process that is affected by environmental factors, while active death can be regulated by genes to prevent death and maintain the internal environment.
Under certain circumstances, autophagy acts as a protector for cells. Literature has shown that autophagy is activated and is basic for maintaining cell survival if growth factors and nutrients are absent in Bax/Bak-deficient mice. 62 However, the authors know that the results of autophagy depend on the type of initiative stimuli, microenvironment, cell types, and other factors. Excessive autophagy tends to cause autophagic cell death. Other than apoptosis, autophagy is known as an alternative form of cell death. 63 Studies in Drosophila have indicated that the steroid hormone causes increased numbers of autophagosome and increased Atg RNAs. 64 Interestingly, midgut destruction is suppressed by impaired Atg1, Atg2, or Atg18, which indicates that autophagy has a vital role in steroid-induced degradation of midget cells. In addition, accumulation of Atg proteins also induces autophagic cell death, which is dependent on Atg5 and Beclin-1, or Bcl-2 family members (Bcl-2 and Bc1-xL) in mammalian cells. 65 Upregulation of Beclin-1 and Atg5 can thereby suppress tumor formation. 66,67 As well, in MCF-7 cells, tamoxifen induces autophagy by the activated class-I PI3K pathway, 68 and TNF-α can stimulate autophagy and apoptosis in T lymphoblastic leukemia cells. 69 However, treatment with the autophagy inhibitor, 3-methyladenine (3-MA), protects these cells from death. More interestingly, this group has discovered excessive autophagy activation in response to IR. For example, ATM increases IR-induced autophagy through the MAPK14 pathway, which contributes to the induction of cell death and the radiosensitization of H1299cells. 70 Moreover, IR can induce autophagic cell death by damaging mitochondria in Hela cells. 6 Inhibition of autophagosome formation with autophagy inhibitors such as 3-MA and Wortmannin, or by the silencing of Atg5 and Atg6 protects cells from death. 6,65
It is now known that oxidative stress is able to regulate autophagy. In HT-29 colon cancer cells, autophagic cell death occurs under oxidative stress in combination with hyperthermia. 71 Moreover, H2O2 can induce autophagic cell death by the BNIP3-mediated suppression of the mTOR pathway. 72 The detailed mechanism will be further described below.
Reactive oxygen species—mitochondrial by-products
ROS have long been considered by-products of cellular metabolism and of organelles such as mitochondria and lysosomes. In particular, mitochondria are major intracellular sources of ROS in mammalian cells, and ROS can participate in various processes, including autophagy. 73 ROS comprised highly active oxygen molecules and their unstable derivatives. Superoxide (O2 −) and hydrogen peroxide (H2O2) are major species of ROS. Both of them have been confirmed to mediate autophagy. 74 Mitochondrial ETC (mETC) complexes I and III contribute mainly to superoxide (O2 −) anion production. 75 O2 − can be catalytically converted to H2O2 by the action of mitochondrial manganese SOD (MnSOD) families, especially SOD1 and SOD2. O2 − can specifically induce autophagy when cell treated with mETC inhibitors (TTFA), the SOD inhibitor 2-methoxyestradiol 76 or exogenous H2O2. 77 –79 ROS participate in multiple autophagy signaling pathways. The autophagy-related gene Beclin-1/Atg6 can be activated by ROS. 80 ROS can also increase AMPK activation and autophagy levels directly or indirectly through the tumor suppressor LKB-1(also known as STK11). In addition, ROS have effects on the activation of p53-DRAM1 pathway and the induction of autophagy. 81 The nuclear high-mobility group box-1protein (HMGB1) can increase autophagy levels in response to ROS by inhibiting mTOR or inducing Beclin-1expression.
The release of ROS from the mitochondrial respiratory chain can lead to increased mitochondrial outer membrane permeability, followed by decreased mitochondrial transmembrane potential and ATP synthesis. Continually increasing ROS causes serious damage to mitochondrial structure and function, and promotes cell aging and even death (Fig. 3). Recently, increasing evidence has indicated the essential roles of ROS in autophagic cell death. For instance, in breast and colon cancer cells, intracellular ROS accumulation can lead to BIX-induced autophagic cell death, 82 while in EW7 cells, inhibition of TNF-α can induce ROS production and trigger ROS-dependent autophagic cell death. 83 Some exogenous drugs, such as the ETC inhibitors, rotenone or TTFA, can induce autophagic cell death via elevated ROS. 78 Furthermore, Chen et al. have demonstrated that exogenous H2O2 treatment can induce autophagic cell death in the transformed cell line HEK293 and the cancer cell lines U87 and Hela. 79 In this group, the role of ROS in autophagic cell death has also been confirmed in Hela cells. 6 After treatment with the antioxidant NAC, autophagy and cell death are both clearly suppressed, suggesting that ROS play an important role in the induction of autophagic cell death.
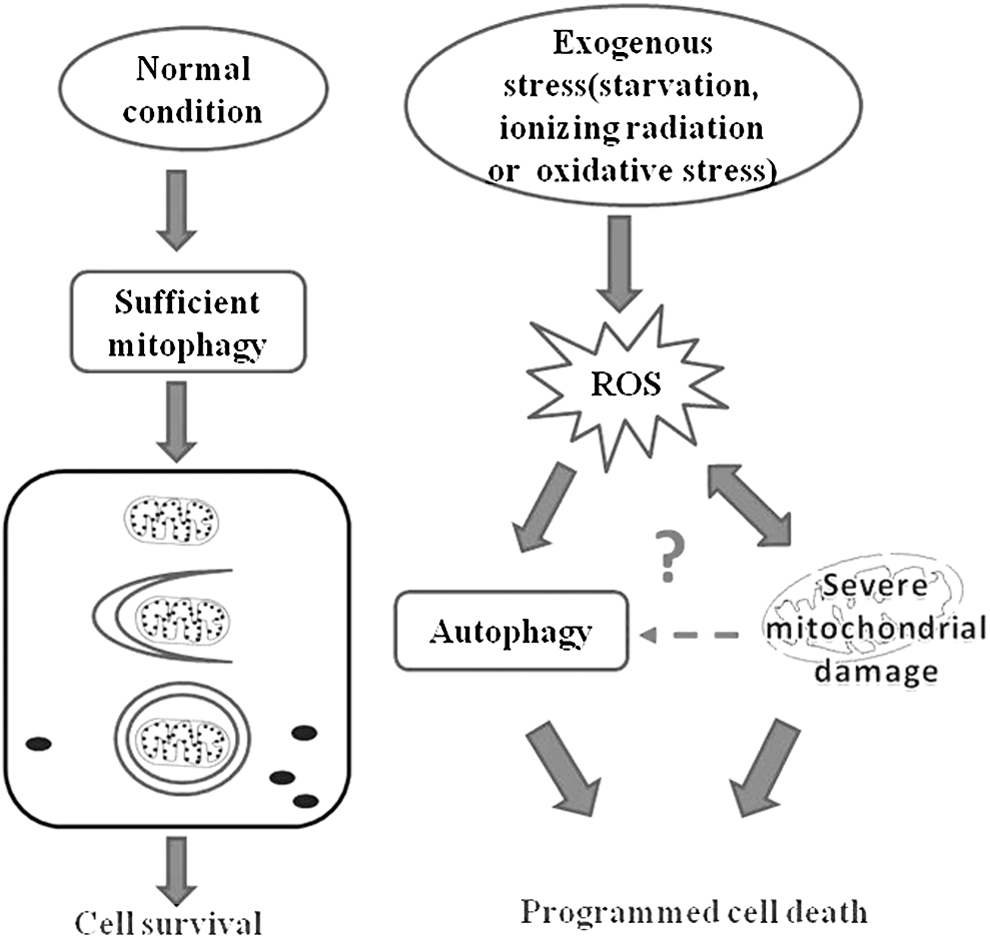
Mitochondria and ROS in cell death. Various studies have shown that ROS levels are significantly increased. Under exogenous stress (starvation, ionizing radiation, or oxidative stress), while mitochondrial damage leads to increased ROS, excessive ROS in turn causes mitochondrial damage. Autophagy induction by ROS is intimately connected to cell death, and mitochondria play a crucial role in determining cell fate. If mitochondrial damage occurs, cells face viability challenges. However, the association between mitochondrial damage and autophagy requires further research. ROS, reactive oxygen species.
The possible mechanisms of mitochondria-mediated autophagy
Mitochondria are ubiquitous organelles in mammalian cells. Recent findings have indicated that the specific inhibition of mitochondrial complexes can have differential effects on autophagy. 84 Rotenone, an environmental pesticide, can potently inhibit complex I and causes mitochondrial dysfunction even at nanomolar concentrations. 85 A number of studies have shown that rotenone can induce autophagy via oxidative stress and increased expression of HIF1-α, and that the level of autophagy can be enhanced by autophagy inducers such as rapamycin. 86 Interestingly, inhibition of complex I with rotenone or inhibition of complex II with 2-thenoyltrifluoroacetone (TTFA) activates autophagy and cell death in HEK293 cells, and the cancer cell lines HeLa and U87. 78 Antimycin A, an inhibitor of complex III, can block autophagy in HeLa cells, but fails to block autophagy in H4 and HeLa cells depleted of mitochondrial DNA. 87 The above studies suggest that mitochondria have an essential role in regulating autophagy.
Mitochondria make significant contributions to both apoptosis and autophagy, two important and interconnected stress–response mechanisms. Studies have shown that full-length Beclin-1 is cleaved into different fragments during IL-3 deprivation, which is mediated by caspases. 88 Although Beclin-1 is an autophagy-associated protein, cleaved Beclin-1 cannot induce autophagy. However, Beclin-1 fragments can be recruited to mitochondria to induce apoptosis. It has also been reported that Beclin-1 and Atg5 can be cleaved on treatment with TNF-related apoptosis-inducing ligand (TRAIL) and in combination with arginine deprivation in melanoma cells. 89 In contrast, inhibition of caspases prevents the cleavage of Beclin-1 and Atg5, and reduces cell death.
Further underscoring the connection between apoptosis and autophagy, some proteins regulate both processes. For instance, protein HMGB1 is a positive regulator of autophagy in leukemia pathogenesis and chemotherapy resistance. 59 When HMGB1 localizes to the membrane of the ER, it contributes to autophagy initiation, while HMGB1 can also participate in induction of apoptosis when it is localized to mitochondria. Similar to HMGB1, autophagy/beclin-1 regulator 1 (AMBRA1) is an autophagy-related protein that regulates both autophagy and apoptosis. In contrast, Bcl-2 can locate on both the ER and mitochondria, where it functions as an inhibitor of autophagy and apoptosis, respectively. 90
Mitochondrial function may also be affected by lysosomes, according to a lysosomal–mitochondrial axis theory proposed by Terman et al. in 2006. 91 The lysosome is an acidic structure that contains several types of hydrolases. Although the lysosome participates in the process of autophagy, impaired lysosomes can also be degraded by autophagy. Various exogenous stimuli, such as the glucoalkaloidsolamargine, 92 may lead to lysosome membrane permeabilization. Lysosomes release many kinds of hydrolases such as cathepsins, 91,93 which consequently participate in the regulation of different types of cell death. Release of these hydrolases may also impair mitochondria, either by lysosomal ROS or by the hydrolase-initiated lysing of mitochondrial outer membrane. The proposal of a lysosomal–mitochondrial axis provides a new approach to study the interaction between autophagy and apoptosis. However, the role of lysosomal–mitochondrial axis in the regulation of cell death still needs further verification.
Conclusions
Autophagy is an important biological process and plays dual roles in cancer development and therapy. 94,95 As one of the most important organelles, mitochondria have special roles in the regulation of autophagy and cell death. Mitochondria regulate autophagy not only via mitochondrial ROS but also by interacting with lysosomes and the ER. Overall, the role of mitochondria in cell death will undoubtedly provide a new target for cancer therapy, along with a new prospect for future application in cancer treatment.
Footnotes
Acknowledgments
The authors thank Miriam Derksen and Alexandra Ciappla for checking grammar and spelling. This study was supported by NSFC grant (31370837 and 81573082), the provincial program of Science and Technology of Jilin (20150101142JC), and the Health and Family Planning Commission of Jilin Province (20142036).
Disclosure Statement
No conflicting financial interests exist.