
Abstract
Background:
Up to now, prostate-specific membrane antigen (PSMA)-targeted radionuclide therapy mainly focused on β-emitting radionuclides. Herein, two new 213Bi-labeled agents for PSMA-targeted α therapy of prostate cancer (PCa) are reported.
Methods:
The biodistribution of 213Bi-labeled small-molecule inhibitor PSMA I&T and nanobody JVZ-008 was evaluated in mice bearing PSMA-positive LNCaP xenografts. DNA damage response was followed using LNCaP cells and LNCaP xenografts.
Results:
In vitro, 213Bi-PSMA I&T and 213Bi-JVZ-008 therapy of LNCaP cells led to increased number of DNA double-strand breaks (DSBs), detected as 53BP1 and γH2AX nuclear foci. In vivo, tumor uptake of 213Bi-PSMA I&T and 213Bi-JVZ-008 was 5.75% ± 2.70%ID/g (injected dose per gram) and 2.68% ± 0.56%ID/g, respectively, with similar tumor-to-kidney ratios. Furthermore, both agents induced in vivo DSBs in the tumors, which were detected between 1 hour and 24 hours after injection. 213Bi-PSMA I&T induced significantly more DSBs than 213Bi-JVZ-008 (p < 0.01).
Conclusions:
213Bi-PSMA I&T and 213Bi-JVZ-008 showed efficient and rapid tumor targeting and produced DSBs in PSMA-expressing LNCaP cells and xenografts. These promising results require further evaluation of 213Bi-labeled agents with regard to their therapeutic efficacy and toxicity for PCa therapy.
Introduction
Late stage metastasized prostate cancer (PCa) is associated with a poor survival and diminished quality of life. Taxane-based therapies and second-line hormonal therapies show only moderate survival benefits and are often associated with toxicity. 1 The recently introduced bone-seeking radiopharmaceutical radium-223 chloride (223RaCl2, Xofigo™) results in reduction of bone pain and overall survival advantage of 3.6 months in castration-resistant PCa patients with bone metastases. 2 However, the use of this promising radiopharmaceutical is limited to the treatment of bone metastases. Therefore, there is an urgent need to develop a PCa-targeted α-therapy approach to target and treat all PCa metastases.
Recent studies have shown that prostate-specific membrane antigen (PSMA) is an important target for radionuclide imaging and therapy of PCa. PSMA is selectively overexpressed in 90%–100% of primary and metastatic PCa lesions. 3,4 Currently, several small-molecule PSMA ligands have made their way into the clinic for PCa imaging (e.g., 68Ga-PSMA-11, 68Ga-PSMA I&T, and 18 F-DCFPyl). 5 –7 Up to now, PSMA-targeted radionuclide therapy mainly focused on β-emitting radionuclides. Studies with small molecule inhibitors and antibodies, such as 131I-MIP-1095, 177Lu-PSMA-617, 177Lu-PSMA I&T, and 177Lu-J591, have shown the safety and therapeutic efficacy of this approach. However, ∼30% of PCa patients do not respond to these agents or develop resistance during treatment. 6,8 –13 The therapeutic efficacy of PSMA-targeted agents could be improved using α-emitting radionuclides. α particles have higher energies (4–9 MeV) and shorter path lengths (40–100 μm) than β particles (energy: 0.1–2.2 MeV, path length: 1–10 mm). The high linear energy transfer (LET) of α particles causes complex DNA double-strand breaks (DSBs), resulting in deletions, chromosome aberrations, and cell death. 14 –16 High LET radionuclides, therefore, have a higher biological effectiveness to kill cancer cells and may significantly enhance the efficacy of radionuclide therapy. 17,18 First in-human studies of PSMA-617 labeled with the long-lived α emitter 225Ac (t1/2 = 9.9 days) have shown remarkable therapeutic efficacy in PCa patients resistant to 177Lu-PSMA or in whom treatment with β emitters was contraindicated because of diffuse red marrow infiltration. 19
Here two PSMA-targeting tracers are evaluated, PSMA I&T and the nanobody JVZ-008, coupled to the α emitter 213Bi. 213Bi (t1/2 = 46 minutes) decays to stable 209Bi with a branching of 97.8% through the emission of a β − particles to 213Po, immediately followed by the emission of the main therapeutically relevant α particle of 8.4 MeV. With a branching of 2.2%, 213Bi decays through the emission of an α particle with 5.9 MeV to 209Tl, followed by the emission of a β −particle to stable 209Bi. 20 PSMA I&T and JVZ-008 are directed against the extracellular domain of PSMA and show rapid accumulation in PSMA-expressing tumors and represent ideal molecular carriers for the short-lived 213Bi. 6,13,21,22 The aim of this study is to assess the tumor targeting and biodistribution profiles of 213Bi-PSMA I&T and 213Bi-JVZ-007-cys-DOTA (213Bi-JVZ-008), as well as the resulting induction of DSBs using the PSMA-expressing human PCa xenograft model LNCaP.
Materials and Methods
All reagents used were purchased from Sigma-Aldrich BV, The Netherlands, unless stated otherwise.
Synthesis and radiolabeling of 213Bi-PSMA I&T and 213Bi-JVZ-008
PSMA I&T was kindly provided by the Technical University Munich and JVZ-007-cys was synthesized as described previously. 6,22 JVZ-008 was obtained by conjugation of JVZ-007-cys to maleimide-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, Chematech, Dijon, France) according to the protocol described for maleimide-maleimide-diethylenetriaminepentaacetic acid (DTPA). 22 213Bi was eluted as BiI4−/BiI52− from a 225Ac/213Bi generator produced by the Directorate for Nuclear Safety and Security (Karlsruhe, Germany), using 600 μL of 0.1 M HCl/0.1 M NaI. For PSMA I&T labeling, the 213Bi eluate was added to a labeling vial containing 120 μL of 2 M Tris, 50 μL of 20% ascorbic acid and 2–4 nmol PSMA I&T (pH 8.7). For JVZ-008 labeling, the 213Bi eluate was added to a labeling vial containing 80 μL of 2 M Tris, 50 μL of 20% ascorbic acid, and 4 nmol JVZ-008 (pH 7.9). The reaction mixture was heated for 10 minutes at 95°C. After the reaction, DTPA was added to a final concentration of 0.03 mM to complex-free 213Bi. Incorporation yield was determined by instant thin-layer chromatography (Agilent Technologies) using 0.1 M NH4OAc/0.1 M EDTA (pH 5.5) and 0.1 M sodium citrate (pH 6.0) as eluent for 213Bi-PSMA I&T and 213Bi-JVZ-008, respectively. Radioactivity corresponding to 213Bi-PSMA I&T (Rf = 0.5),213Bi-JVZ-008 (Rf = 0), and free 213Bi (Rf = 1) was quantified using a gamma counter (Perkin Elmer). Incorporation yield always exceeded 95%.
For animal experiments, radiolabeled tracers were purified and formulated in a physiological buffer. 213Bi-PSMA I&T was purified using an OasisTM HLB column (Waters, Milford, MA) preconditioned with 1 mL ethanol and 2 mL water. 213Bi-PSMA I&T was loaded on the column and eluted with 500 μL ethanol. The volume was reduced to 50 μL by evaporation, 50 nmol 2-(phosphonomethyl)pentane-1,5-dioic acid (PMPA; Santa Cruz Biotechnology, Dallas, TX) was added and phosphate-buffered saline (PBS)/0.5% bovine serum albumin (BSA) was added up to 1 mL. Mice received 200 μL of this solution. 213Bi-JVZ-008 was purified by size exclusion chromatography using an illustra NAP-5 column (GE Healthcare Life Sciences, The Netherlands) preconditioned with 5 volumes of 0.5% BSA and eluted with 1 mL PBS in 100 μL fractions. The first five fractions were pooled and mice were coinjected with 100 μL of this solution combined with 100 μL gelofusine (40 mg/mL). Specific activity was 30 MBq/nmol and 7 MBq/nmol at time of injection for 213Bi-PSMA I&T and 213Bi-JVZ-008, respectively.
Cell culture
The human PCa cell line LNCaP was cultured in RPMI1640 (GIBCO, BRL Life Sciences Technologies, The Netherlands), with 2 mM glutamine (GIBCO) and 10% fetal calf serum (at 37°C with 5% CO2).
In vitro experiments
LNCaP cells were seeded in 12-well plates (5 × 104 cells in 1 mL/well) on sterile 13-mm diameter cover slips and allowed to grow for 2 days. Subsequently, cells were treated for 20 minutes at 37°C with 300 μL of 213Bi-PSMA I&T (10−7 M, 0.3 MBq), 213Bi-JVZ-008 (10−7 M, 0.3 MBq), nontargeted tracer 213Bi-DTPA (2 × 10−6 M, 0.3 MBq), or medium containing labeling buffer. Immediately after treatment, cells were washed with 1 mL PBS and incubated with normal medium for 1, 2, 4, 24, or 48 hours. After incubation, cells were washed with 2 mL PBS, fixed for 15 minutes in 0.5 mL of 2% paraformaldehyde at room temperature (RT), and washed with 1 mL PBS.
In vivo experiments
All animal experiments were conducted in accordance with the revised Dutch Act on Animal Experimentation (1997) and approved by the institutional Animal Welfare Committee of the Radboud University Nijmegen.
Female nude BALB/c mice (4 animals per condition, age 6–8 weeks; Janvier Lab, Le Genest-Saint-Isle, France) were inoculated subcutaneously with LNCaP cells (3 × 106 cells, 200 μL, 33% RPMI/67% matrigel, BD Biosciences, Pharmingen, San. Diego, CA). Three weeks after inoculation (tumors 4–5 mm in diameter), 213Bi-PSMA I&T (0.2 nmol, 5.4–6.6 MBq) was injected intravenously with 10 nmol 2-PMPA for renal protection and 213Bi-JVZ-008 (0.7 nmol, 4.5–5.4 MBq) was injected intravenously with 4 mg gelofusine for renal protection. 21,23 Nontreated animals were taken along as controls.
At 1 and 24 hours after injection, mice were euthanized using CO2/O2 asphyxiation and tumor, blood, muscle, lung, spleen, kidney, liver, small intestine, colon, and salivary glands were collected and weighed. Tumors were fixed in 4% paraformaldehyde. For biodistribution studies at 1 hour after injection, radioactivity in each organ was determined in a γ counter by measuring the γ emission of 213Bi (440 keV), and the percentage of the injected dose per gram (%ID/g) was calculated. After radioactive decay (>10 half-lives), fixed tissues were embedded in paraffin.
Immunofluorescence staining cells
Phosphorylation of histone H2AX (γH2AX) and accumulation of p53-binding protein 1 (53BP1) at the site of DSBs are biomarkers of radiation-induced DSBs. 24 Fixed LNCaP cells were permeabilized for 20 minutes at RT in PBS containing 0.1% Triton X-100 and incubated in blocking buffer (PBS, 0.1% Triton X-100, 2% BSA) for 30 minutes at RT. Next, cells were stained for 90 minutes at RT with primary antibodies [anti-53BP1 (NB100-304, dilution 1/1000; Novus Biologicals, Littleton, CO) and anti-γH2AX (05-636, dilution 1/500; Merck-Millipore, Billerica, Massachusetts)] diluted in blocking buffer. After incubation, cells were washed with PBS 0.1% Triton X-100 and incubated with the secondary antibody (goat antirabbit Alexa Fluor 594 or goat antimouse Alexa Fluor 488, 1/1000; Thermo Fisher Scientific, Waltham, MA) in blocking buffer for 60 minutes at RT. Cells were washed with PBS and mounted with Vectashield containing DAPI (Vector Laboratories, Burlingame, CA). Fluorescent Z-stack imaging was performed using a TCS SP5 confocal microscope (Leica, Wetzlar, Germany).
Immunofluorescence staining tumor tissue
For analysis of in vivo DSBs induction, tumors were paraffin embedded. Four micrometer tissue sections were deparaffinized in xylene and subsequently rehydrated by incubation in decreasing concentrations of ethanol. Target antigen retrieval was performed using a Target Retrieval Solution pH6.1 (Agilent Technologies, Santa Clara, CA) for 18 minutes in a microwave at 650 W. Immunofluorescent stainings and imaging were performed as described for cells. Pictures were processed using the Temporal-Color Code mode from Image J software. Manual α tracks quantification of on average 375 cells of 4 animals per condition was performed. Foci are considered to form an α track when they are observed as a line or when at least four foci lay in the same line within a nucleus.
Statistical analysis
Statistical analyses were performed using PASW Statistics version 22.0 (Chicago, IL). Differences in tracer uptake were tested for significance using the nonparametric Mann–Whitney U test. Differences in number of α tracks were determined using a homoscedastic two-tailed Student's t-test. Samples were considered statistically different if p < 0.05 with **p < 0.01 and ***p < 0.001.
Results
213Bi-radiolabeling of PSMA I&T and JVZ-008
The labeling procedure is depicted in Supplementary Figure S1 (Supplementary Data are available online at
213Bi-PSMA I&T and 213Bi-JVZ-008 induce DSBs in LNCaP cells in vitro
Therapy with the PSMA-targeted tracers 213Bi-PSMA I&T and 213Bi-JVZ-008 and the nontargeted tracer 213Bi-DTPA all induced DSBs in LNCaP cells, as shown by the presence of 53BP1 and γH2AX foci (Figure 1 and Supplementary Fig. S2). At early time points, both single foci and linear tracks, that is, α tracks, were visible. Tracks were present at least until 4 hours after treatment, but were not observed anymore at 24 and 48 hours after treatment. Single DSB foci were still present at these later time points. Background 53BP1 and γH2AX nuclear foci, most likely arising from replication stress, were observed in vehicle-treated LNCaP cells.
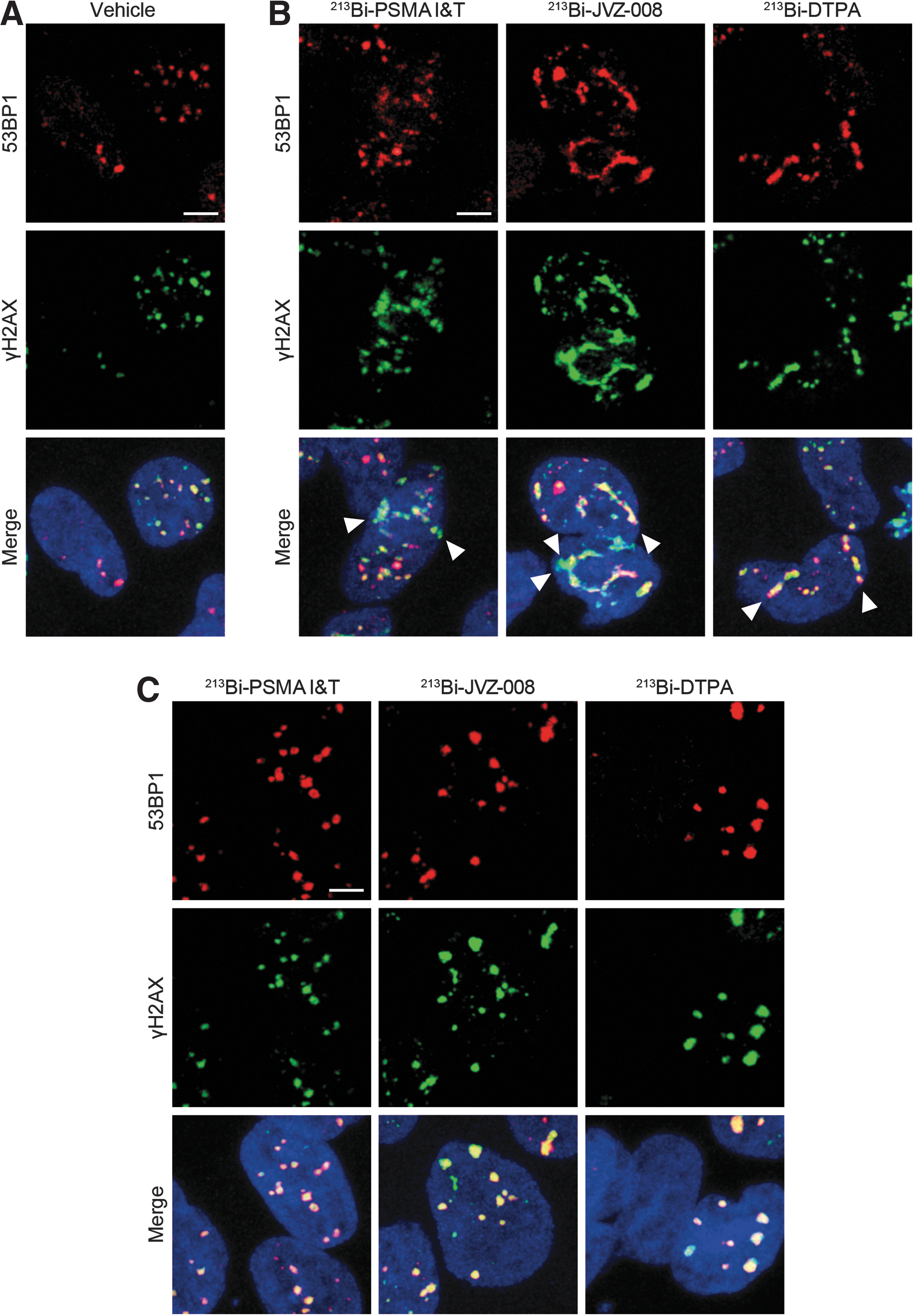
Representative pictures of DNA double-strand breaks (visualized by immunofluorescent staining of γH2AX and 53BP1) in LNCaP cells at 1 hour after treatment with vehicle
213Bi-PSMA I&T and 213Bi-JVZ-008 accumulate in LNCaP tumors in mice
213Bi-PSMA I&T and 213Bi-JVZ-008 accumulated in LNCaP tumors and in kidneys (Fig. 2). Tumor uptake was 5.75% ± 2.70%ID/g and 2.68% ± 0.56%ID/g for 213Bi-PSMA I&T and 213Bi-JVZ-008, respectively (1 hour after injection, p = 0.057). Both tracers cleared rapidly from blood (213Bi-PSMA I&T: 0.30% ± 0.21%ID/g and 213Bi-JVZ-008: 0.30% ± 0.06%ID/g). 213Bi-PSMA I&T showed higher kidney uptake than 213Bi-JVZ-008 (p = 0.029). However, tumor-to-kidney ratios of both compounds were similar (213Bi-PSMA I&T: 0.19 ± 0.10 and 213Bi-JVZ-008: 0.17 ± 0.01). Furthermore, uptake of 213Bi-PSMA I&T was observed in the spleen (2.12% ± 0.23%ID/g).
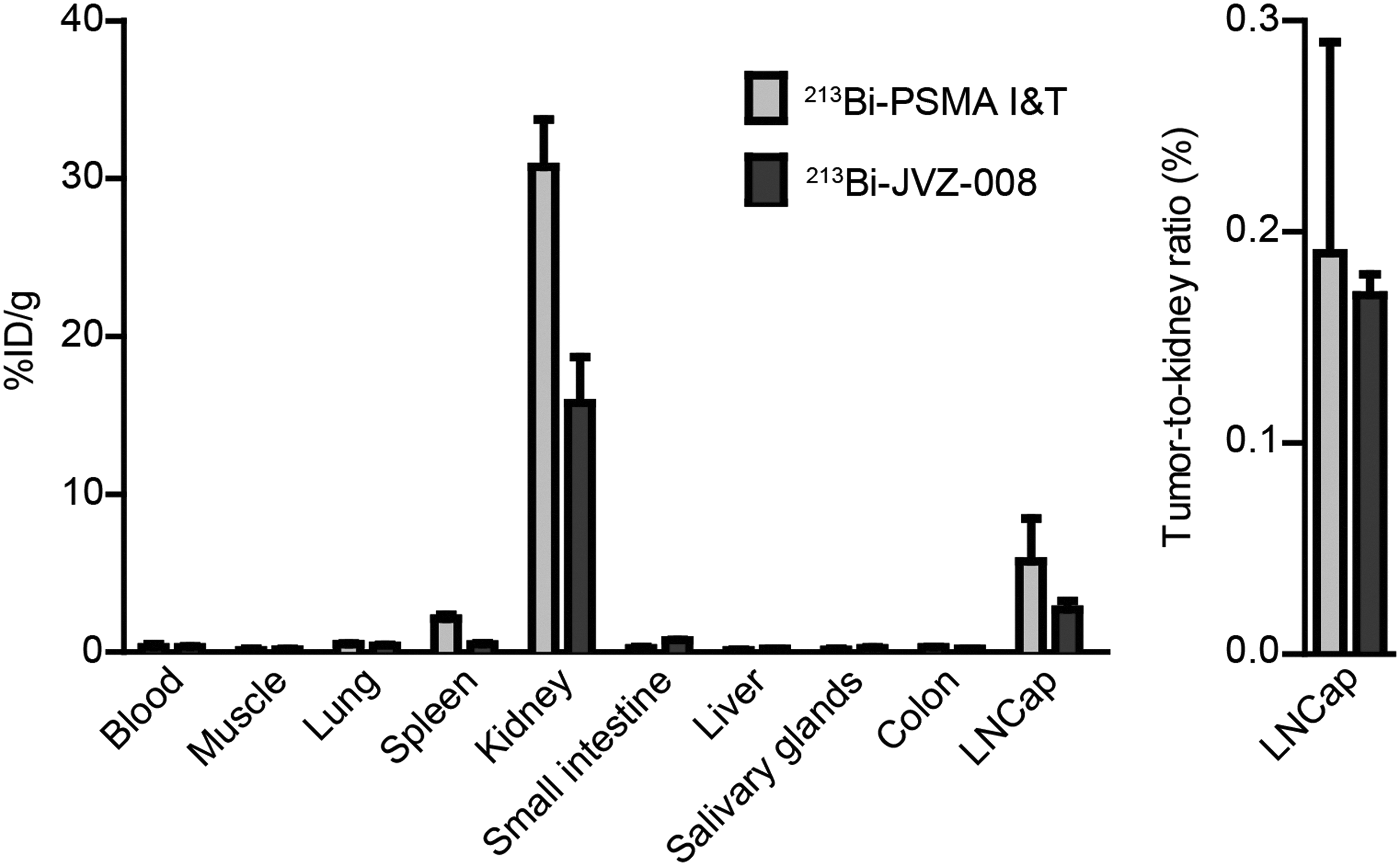
Ex vivo biodistribution of 213Bi-PSMA I&T and 213Bi-JVZ-008 in LNCaP xenografts at 1 hour after injection. Uptake was measured as percentage of injected dose per gram (%ID/g). Error bars represent the SD. PSMA, prostate-specific membrane antigen; SD, standard deviation.
213Bi-PSMA I&T and 213Bi-JVZ-008 induce DSBs in LNCaP tumors in mice
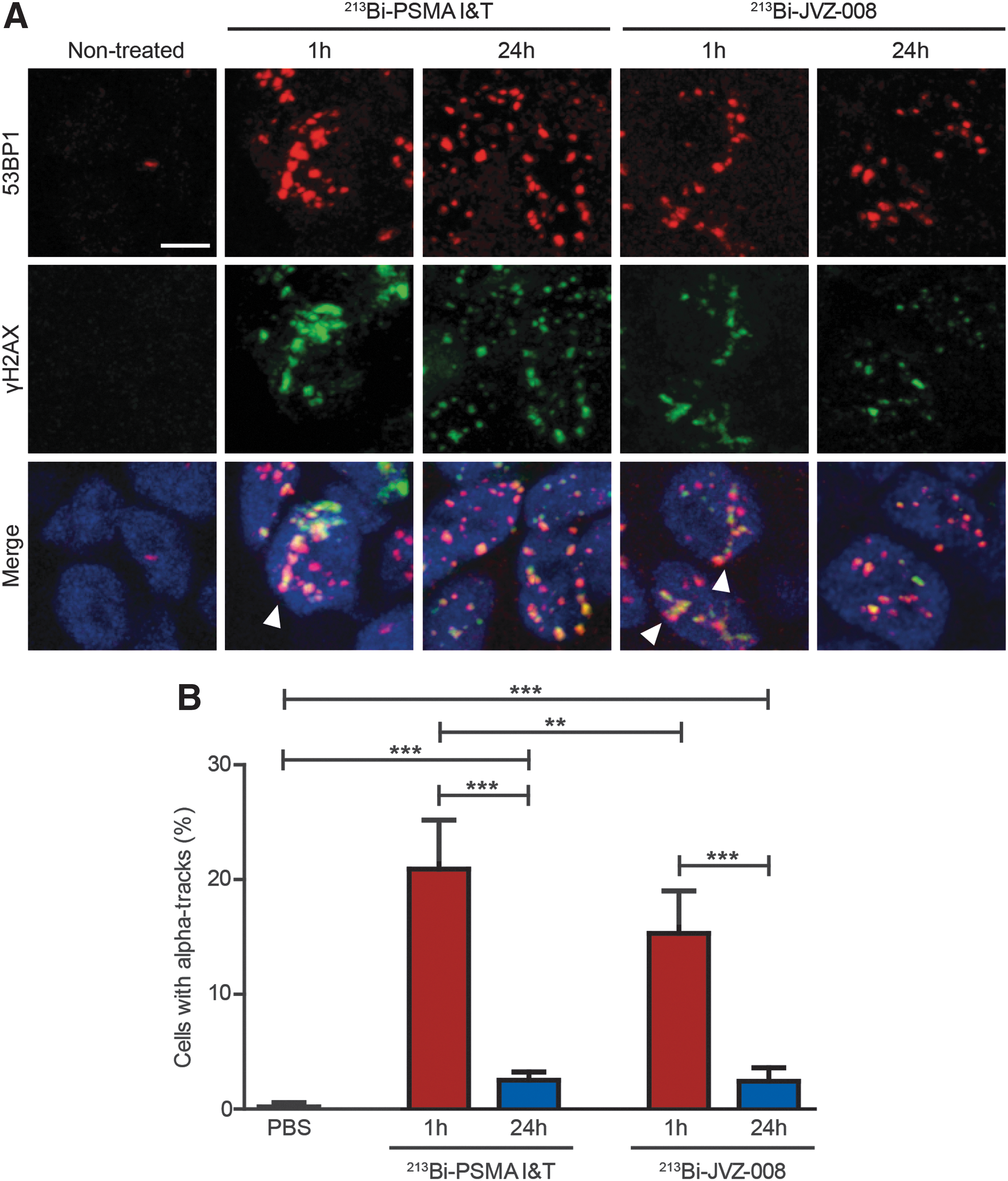
DSBs were induced in LNCaP tumors by both 213Bi-PSMA I&T and 213Bi-JVZ-008, as reflected by increased number of 53BP1 and γH2AX foci 1 hour after injection, compared with nontreated mice (Fig. 3A and Supplementary Fig. S3). A large fraction of cells with α tracks were observed shortly after treatment; in 213Bi-PSMA I&T-treated animals, significantly more α tracks in the tumors (20.91% ± 1.23%) were observed than in 213Bi-JVZ-008-treated animals (15.30% ± 1.07%) (Fig. 3B). Most of the α tracks disappeared 24 hours after injection, but a small portion of the cells still contained these tracks (2.51% ± 0.21% for 213Bi-PSMA I&T and 2.43% ± 0.34% for 213Bi-JVZ-008). Moreover, separate DSBs were still observed 24 hours after injection.
Discussion
Here, the preclinical results focusing on DNA damage induction by PSMA-targeted α radionuclide therapy with two novel PSMA-targeting agents are reported: the small-molecule PSMA inhibitor 213Bi-PSMA I&T and the anti-PSMA nanobody 213Bi-JVZ-008. 213Bi-PSMA I&T and 213Bi-JVZ-008 showed rapid and efficient tumor targeting and produced DSBs in PSMA-positive LNCaP tumors in vivo.
Recently, several other preclinical studies have reported on PSMA-targeted radionuclide therapy using α-emitting radionuclides. In vitro and in vivo treatment with the monoclonal antibody J591 labeled with 213Bi showed promising results. 25,26 However, in vivo application of this tracer appeared challenging because the short 213Bi half-life does not match with slow antibody pharmacokinetics. Two studies showed that anti-PSMA-targeted liposomes/lipid vesicles loaded with the α emitter 225Ac can kill PSMA-expressing cells in vitro. 27,28 However, because of the high recoil energy, Ac-225 daughter products are released from the carrier. Therefore, stability and normal tissue toxicity of these radiotracers should be addressed. 29 A study by Kiess et al. showed the potential of 211At-6 in vitro and in vivo treatment, 30 but 211At was reported to lose therapeutic efficacy with increasing tumor size. 31 Therefore, an optimal combination between a short-lived α emitter and PSMA ligand to induce DNA damage is explored. PSMA I&T and JVZ-008 show rapid accumulation in PSMA-expressing tumors (<1 hour) and, therefore, represent ideal molecular carriers for the short-lived 213Bi. In this study, for the first time the application of 213Bi with these low-molecular weight PSMA-targeting agents is shown.
213Bi-PSMA I&T and 213Bi-JVZ-008 therapy of LNCaP cells resulted in production of DSBs, both in vitro and in vivo. However, induction of DSBs was also observed in vitro after treatment with the nontargeted agent 213Bi-DTPA, with similar DSB repair kinetics as 213Bi-PSMA I&T and 213Bi-JVZ-008. This suggests that DNA damage was induced by 213Bi from the medium during the 20 minutes incubation period. Because of the high cytotoxicity of 213Bi and its short half-life, this in vitro model appeared suboptimal and the tracers were further evaluated in vivo.
In the xenograft model, a twofold higher tumor uptake was observed for 213Bi-PSMA I&T than for 213Bi-JVZ-008 with a similar tumor-to-kidney ratio. The biodistribution of 213Bi-PSMA I&T and 213Bi-JVZ-008 was comparable with that of 111In- and 177Lu-labeled tracers. 21,22 213Bi-PSMA I&T showed significantly higher uptake in the kidneys than 213Bi-JVZ-008. Both tracers are cleared through the kidneys. However, the uptake of 213Bi-PSMA I&T seems to be PSMA mediated, because it can be blocked by coinjecting an excess of unlabeled PSMA I&T or 2-PMPA, and not by gelofusin. 21 In contrast, 213Bi-JVZ-008 uptake can be blocked by coinjecting gelofusin. 22 So far, no studies have been carried out to assess the effect of coinjection of 2-PMPA or an excess of unlabeled JVZ-008.
The higher uptake of 213Bi-PSMA I&T in the tumor correlated with a significant increase in the number of cells with α tracks after 213Bi-PSMA I&T compared with 213Bi-JVZ-008. The number of cells with α tracks was significantly reduced at 24 hours after treatment for both tracers, indicating that a fraction of the DSBs in the tracks was repaired and/or that the chromatin was rearranged. These first results indicate that 213Bi-PSMA I&T is more potent to induce DNA damage in the tumor, but because kidney uptake is also enhanced, nephrotoxicity needs to be monitored closely in the future.
For clinical translation, it is important to note that PSMA is also expressed in normal tissues, and dosimetric calculations for β emitters predict the highest absorbed doses in salivary glands and kidneys. 8,32 So far, only one study reported on clinical use of PSMA-targeted α therapy. In 2 PCa patients, 225Ac-PSMA-617 induced prostate-specific antigen decline and complete imaging response while no relevant hematological toxicity was observed. However, patients did report moderate to severe xerostomia. 19 Furthermore, 213Bi-labeled DOTATOC has been shown to be effective in patients with neuroendocrine tumors refractory to 90Y- and 177Lu-DOTATOC. Although the safety profile was acceptable, patients experienced moderate acute hematological toxicity and moderate chronic kidney toxicity. 33
In conclusion, 213Bi-PSMA I&T and 213Bi-JVZ-008 showed efficient and rapid tumor targeting and produced DSBs in PSMA-expressing LNCaP tumors. These results require further evaluation of therapeutic efficacy and toxicity. Potentially, these novel tracers could offer a promising new treatment option for PCa patients with metastatic lesions.
Footnotes
Acknowledgments
The authors thank Bianca Lemmers-van de Weem, Iris Lamers-Elemans, Kitty Lemmens-Hermans, and Sharon Wennekers for their assistance during animal experiments. Fluorescent imaging was performed in collaboration with the optical imaging center of the Erasmus MC. This study was supported by the Erasmus MC grant “Novel Radio-Antagonists for PET/MRI Imaging and Therapy of Prostate Cancer” and The Netherlands Organization for Scientific Research (ZON-MW grant 40-42600-98-018).
Disclosure Statement
No conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.