
Abstract
Background:
Sorafenib is a multikinase inhibitor that has been approved for the treatment of patients with advanced 131iodine (131I) refractory differentiated thyroid cancer (DTC). However, the progression-free survival of patients with advanced 131I refractory DTC is short, and most DTC patients eventually acquire resistance to sorafenib. Therefore, new therapeutic strategies need to be developed.
Materials and Methods:
The thyroid cancer cell lines 8505C and FTC133 were treated with sorafenib in the presence or absence of BEZ235 or small interfering RNA (siRNA) directed against AKT. A CCK8 kit was used to evaluate cell viability. Protein expression levels of relevant genes were determined by Western blotting analysis, whereas messenger RNA expression levels were determined by real-time PCR analysis. Flow cytometry was performed to assess the number of apoptotic cells.
Results:
The results indicate that sorafenib simultaneously inhibited the activities of the MAPK and PI3K/AKT/mTOR pathways in thyroid cancer cells. Treatment of 8505C and FTC133 cells with NVP-BEZ235, siRNA against AKT, or sorafenib induced tumor cell apoptosis and led to reduced tumor cell proliferation. Sorafenib in combination with PI3K/AKT/mTOR inhibition by NVP-BEZ235 or AKT siRNA enhanced apoptosis and proliferation suppression.
Conclusions:
The evidence of this study suggests that a combinatorial approach that inhibits both the MAPK and PI3K/AKT/mTOR pathways exerts a greater antitumor effect than sorafenib alone in thyroid cancer cell lines.
Introduction
The incidence of thyroid cancer has increased sharply in China. 1 Differentiated thyroid cancer (DTC) accounts for ∼95% of all thyroid cancers worldwide. 2 131Iodine (131I) is an effective treatment measure for distant metastases of DTC after total thyroidectomy. However, >50%–70% of distant metastases in DTC cases eventually become refractory to 131I. 3,4 Therefore, limited methods exist for the treatment of 131I-resistant cases because radiation and chemotherapy are usually not effective.
Sorafenib is a multiple kinase inhibitor that has been verified to target vascular endothelial growth factor (VEGF) receptor-1, −2, and −3, platelet-derived growth factor (PDGF) receptor-beta, B-RAF, RET, c-kit, and Flt-3; this drug is also involved in apoptosis, autophagy, antiproliferation, and inhibition of angiogenesis regulation. 5 –7 Sorafenib was approved for treatment of advanced and progressive radioactive iodine-refractory DTCs. A phase 3 clinical trial demonstrated that sorafenib significantly improved progression-free survival (PFS) compared with placebo in patients with advanced/progressive radioactive iodine-refractory DTC. 5,7 However, most patients acquired resistance after 1 or 2 years of sorafenib administration, and the potential mechanism for this acquired resistance needs to be further demonstrated. 5
Cancers are thought to be promoted or driven by damaged genes or by the abnormal activation of oncogene pathways. 8 More than 70% of DTC patients have tumors with MAPK pathway activation because of BRAF and RAS mutations. 9 Copy number gains in the PI3K/AKT pathway are also important for the progression and aggressiveness of thyroid cancer. 10 Although RAS is thought to be a dual activator of both the MAPK and PI3K/AKT pathways, it preferentially activates the PI3K/AKT pathway in DTC. 9,10 Several studies have indicated that sorafenib reduces the phosphorylation of mTORC1, both in vivo and in vitro. 11 –13
However, the phosphorylation levels of AKT are either increased or decreased by sorafenib, which is the opposite of the finding already described. 11 –14 The potential mechanism for this may be that exposure to sorafenib activates AKT through the feedback loop of mTOR. 13 Both MAPK and PI3K/AKT pathway activities enhance the malignant phenotype of thyroid cancer and cause resistance to anticancer therapy. 9,10 A clinical trial indicated that a lower level of nuclear pAKT expression was associated with a higher rate of response to sorafenib. 15
The PI3K/AKT pathway is closely related to the outcomes of sorafenib treatment in patients with DTC. However, the relationship between the PI3K/AKT pathway and sorafenib treatment for DTC currently lacks evidence. This work is the first to demonstrate the role of the PI3K/AKT pathway in sorafenib treatment for DTC and the interaction between this pathway and the treatment mechanism. However, the depth of this mechanism and its significance in clinical practice should be further investigated.
Materials and Methods
Cell lines and reagents
The human thyroid cancer cell lines 8505C and FTC133 were cultured at 37°C in Dulbecco's modified Eagle medium (GIBCO-ThermoFisher, Shanghai, China) supplemented with 10% fetal bovine serum (GIBCO-ThermoFisher), 100 U/mL penicillin, and 100 g/mL streptomycin in a humidified incubator with 5% carbon dioxide (CO2). Antibodies to GAPDH, p-ERK, ERK, and AKT were purchased from Santa Cruz (Texas), and the anti-p-AKT (Ser473) antibody was purchased from Abcam (Cambridge, UK). The p70s6 and p-p70s6 antibodies were purchased from Cell Signaling Technology (Danvers, MA). Sorafenib was generously given to us by the Bayer Company and BEZ-234 was obtained from Selleck (Shanghai, China). Cells were transfected with scrambled small interfering RNA (negative control siRNA) or AKT siRNA (GenePharma, Shanghai, China) using Lipofectamine™ 2000 (Invitrogen-ThermoFisher). The siRNA sequences are provided in the Supplementary Table S1 (Supplementary Data are available online at
Western blotting analysis
Cell pellets were lysed in Radio-immuno precipitation assay (RIPA) lysis buffer and a protease inhibitor cocktail (Sigma, Shanghai, China). Next, 30 or 60 μg of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and analyzed by immunoblotting using chemiluminescence (Santa Cruz). Blots were incubated in 5% milk for 2 hours at room temperature to block nonspecific antibody binding. Blots were later incubated overnight at 4°C with antibodies against the target proteins. A mouse antigoat or antirabbit antibody (Santa Cruz) was diluted 1:1000 in 5% skim milk, Tris-HCl (pH 7.5), and 0.1% Tween-20. The immunoblots were subsequently washed and incubated with the mouse antigoat or antirabbit antibody.
RNA extraction and quantitative real-time PCR
Total RNA was isolated using RNA prep Pure Kit according to the manufacturer's instructions (TIANGEN). The synthesis of complementary DNA by reverse transcription was performed with HiScript II Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech, Nanjing, China). PCR was performed with anAceQ qPCR SYBR Green Master Mix (without ROX) (Vazyme Biotech, Nanjing, China). GAPDH was used as a quality control. Primer sequences used in the real-time PCR analysis are provided in the Supplementary Table S2.
Apoptosis assay
Cells (2 × 105/well) were seeded in a six-well plate. After treatment, the cells were harvested and analyzed for apoptosis by Annexin-V and propidium iodide staining, using an FITC Annexin-V apoptosis detection kit (Life Technologies) according to the manufacturer's instructions and a flow cytometer (FACS; Beckman Coulter, Miami, FL).
Cell proliferation and viability assays
Cells were plated at a density of 1 × 104 cells per well in 96-well microtiter plates, and each plate was incubated for 24 hours at 37°C in 5% CO2. After treatment, the absorbance of the contents of each well was measured at 470 nm with a CCK8 kit (Beyotime, Guangzhou, China).
Statistical analysis
Statistical evaluations are presented as the mean ± standard error. Data were analyzed using Student's t-test. p-Values were considered to be significant if p < 0.05.
Results
Sorafenib effectively inhibits the proliferation of thyroid cancer cells
8505C and FTC133 cells were treated with various concentrations of sorafenib (0.3125 to 80 μM), and a CCK8 assay was used to evaluate proliferation. As indicated in Figure 1A, sorafenib suppresses thyroid cell line proliferation in a dose- and time-dependent manner. A concentration of sorafenib >5 μM inhibited the proliferation of both 8505C and FTC133 cells. This inhibition was greater as time progressed (24, 48, and 72 hours) (Fig. 1A). After incubation with 10 μM sorafenib for 48 hours, the viability rates of 8505C and FTC133 cells were 66.4% and 80.1%, respectively. When the concentration of sorafenib reached 20 μM and the cells were incubated with sorafenib for 72 hours, the viability rates of 8505C and FTC133 cells were 54.2% and 60.2%, respectively. Flow cytometry was used to evaluate cellular apoptosis after sorafenib treatment (Fig. 1B, C). The results indicate that apoptosis increased after treatment of the cells with 5 μM sorafenib for 72 hours. After incubation with 10 μM sorafenib for 72 hours, the rate of apoptosis of 8505C and FTC133 cells increased to 22.9% and 56.7%, respectively.
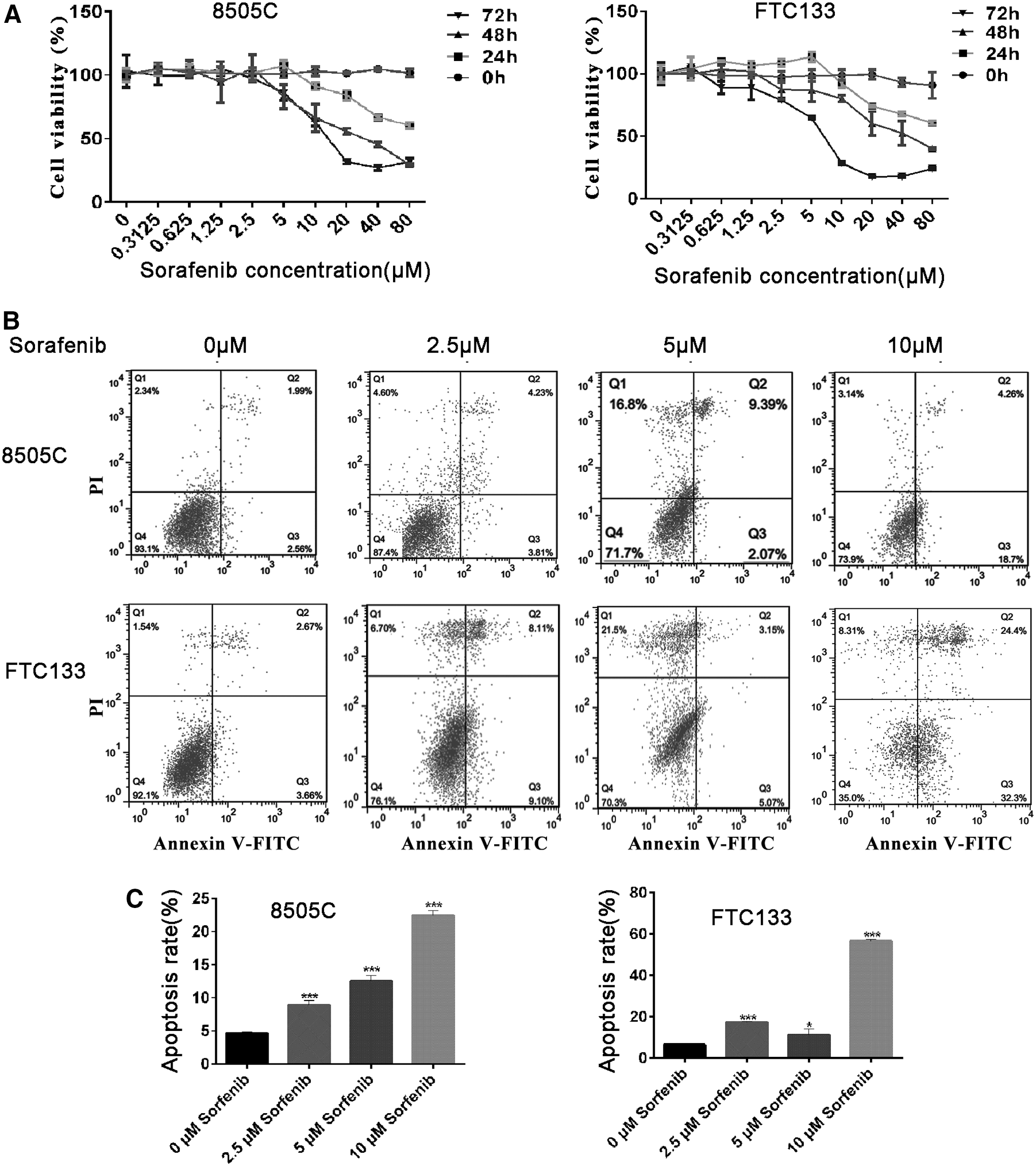
Sorafenib induced cells death and apoptosis in a dose dependent manner.
Sorafenib induces both MAPK and AKT/mTOR pathway inhibition
After incubation of 8505C and FTC133 cell lines with 2.5, 5, or 10 μM sorafenib for 24 hours, the expression of ERK was similar to that of control (Fig. 2A). However, the level of phosphorylated ERK decreased after treatment with 5 and 10 μM sorafenib for 24 hours, but 10 μM sorafenib inhibited the phosphorylation of ERK more strongly (Fig. 2A). Moreover, sorafenib played a similar role in the regulation of AKT and the expression of phosphorylated AKT. The expression of phosphorylated AKT was significantly suppressed after incubation of the cells with 10 μM sorafenib for 24 hours (Fig. 2B). p70s6k is a downstream factor of AKT, and the expression level of phosphorylated p70s6k was significantly reduced after the cells were incubated with 10 μM sorafenib for 24 hours (Fig. 2B). These results indicate that sorafenib inhibited both the ERK and the AKT/mTOR pathway.
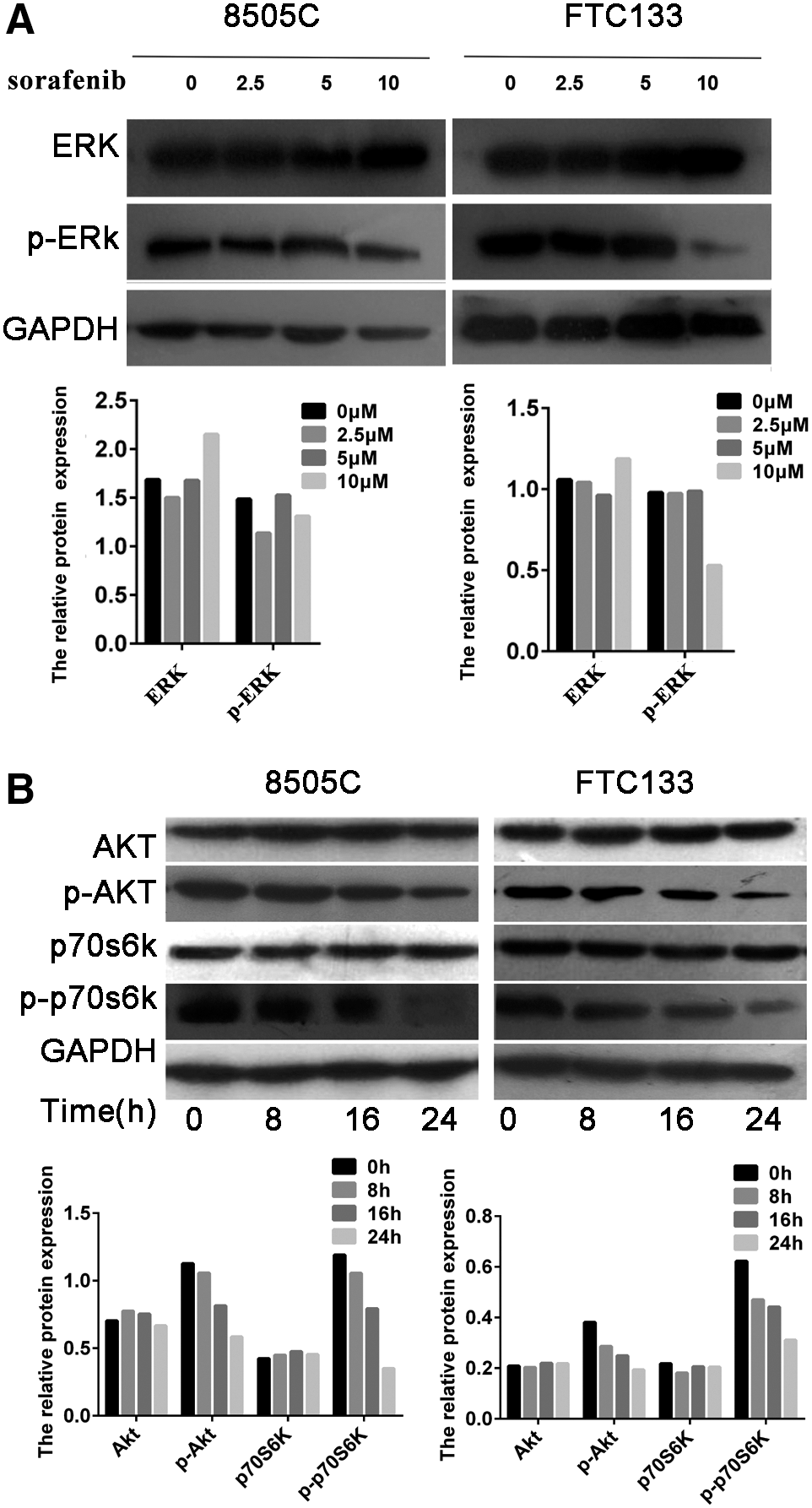
Sorafenib inhibited phosphorylation of ERK and key enzymes in the PI3K/AKT/mTOR pathway.
Silencing of AKT promotes apoptosis and potentiates the therapeutic effects of sorafenib
The role of AKT in sorafenib treatment of thyroid cancer was next demonstrated. Three AKT messenger RNA (mRNA) interference mimics were used to silence AKT mRNA expression. siRNA2 performed the highest with respect to its ability to efficiently silence AKT mRNA and was, therefore, selected for further use (Fig. 3A, B). The silencing of AKT by siRNA2 reduced the expression of phosphorylated AKT and phosphorylated p70s6k protein. The silencing of AKT combined with sorafenib treatment reduced the expression of phosphorylated AKT and phosphorylated p70s6k protein to a greater extent than AKT siRNA2 alone (Fig. 3C).
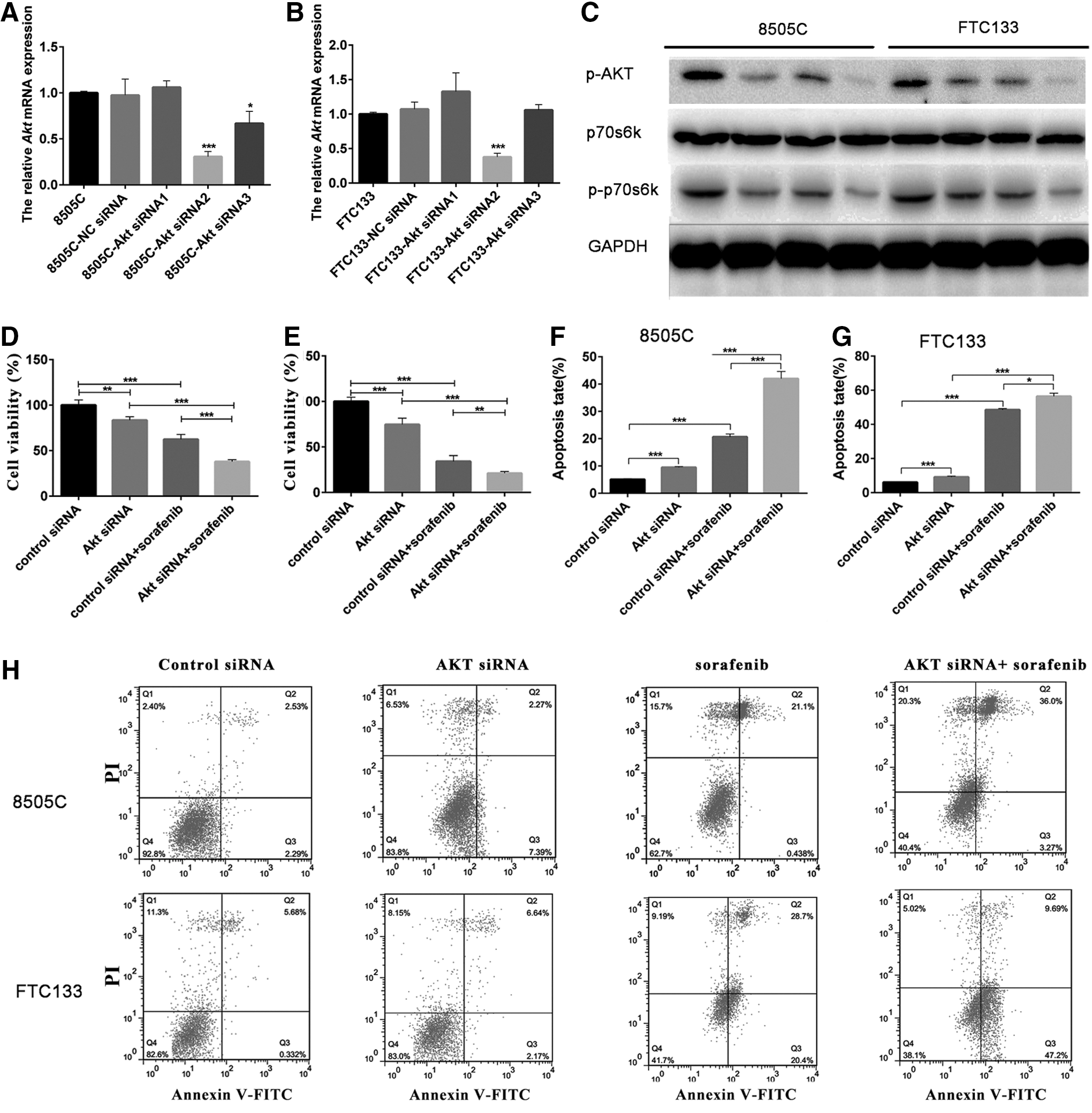
Knockdown AKT mRNA expression promoted apoptosis and therapeutic effects induced by sorafenib.
As shown in Figure 3D and E, the silencing of AKT suppressed the proliferation of 8505C and FTC133 cells. After the disruption of AKT mRNA for 48 hours by siRNA2, the viability rates of 8505C and FTC133 cells were determined to be 83.5% and 74.7%, respectively. Furthermore, treatment of 8505C and FTC133 cells with AKT siRNA2 combined with sorafenib enhanced cell death (Fig. 3D, E). After combination treatment, the cell viability rates of 8505C and FTC133 were 40.0% and 21.1%, respectively. After AKT silencing or sorafenib treatment for 48 hours, the rates of apoptosis of 8505C cells were 9.6% and 21.5%, respectively (Fig. 3F, H). The combined effects of AKT silencing and sorafenib treatment improved the rate of apoptosis to 39.2% in the 8505C cell line (Fig. 3F, H). After AKT silencing or sorafenib treatment for 48 hours, the rates of apoptosis of FTC133 cells were 8.8% and 49.1%, respectively (Fig. 3G, H). The combined effects of AKT silencing and sorafenib improved the rates of apoptosis of FTC133 cells to 56.8% (Fig. 3G, H).
Combination of BEZ235 and sorafenib effectively enhances thyroid cancer therapy
As a dual inhibitor of PI3K and mTOR, BEZ235 effectively inactivates the PI3K/AKT/mTOR pathway and its downstream effectors. After incubation with 0.2 μM BEZ235 for 72 hours, the viability of 8505C and FTC133 cells was suppressed (Supplementary Fig. S1). No significant difference was observed in the sensitivity of 8505C and FTC133 cells. When the concentration of BEZ235 was established at 0.20, 0.4, and 0.8 μM, the cells were incubated for 72 hours, and the viability rates of 8505C cells were 57.7%, 71.2%, and 79.4%, respectively, whereas those for FTC133 cells were 45.8%, 53.4%, and 65.3%, respectively (Supplementary Fig. S1). When the cells were incubated with BEZ235 for 24 hours, 0.4 and 0.8 μM BEZ235 reduced the phosphorylation levels of AKT and p70s6k, but the effects of 0.8 μM BEZ235 were greater (Fig. 4A, B). The combination of BEZ235 and sorafenib enhanced the dephosphorylation of p70s6k (Fig. 4B).
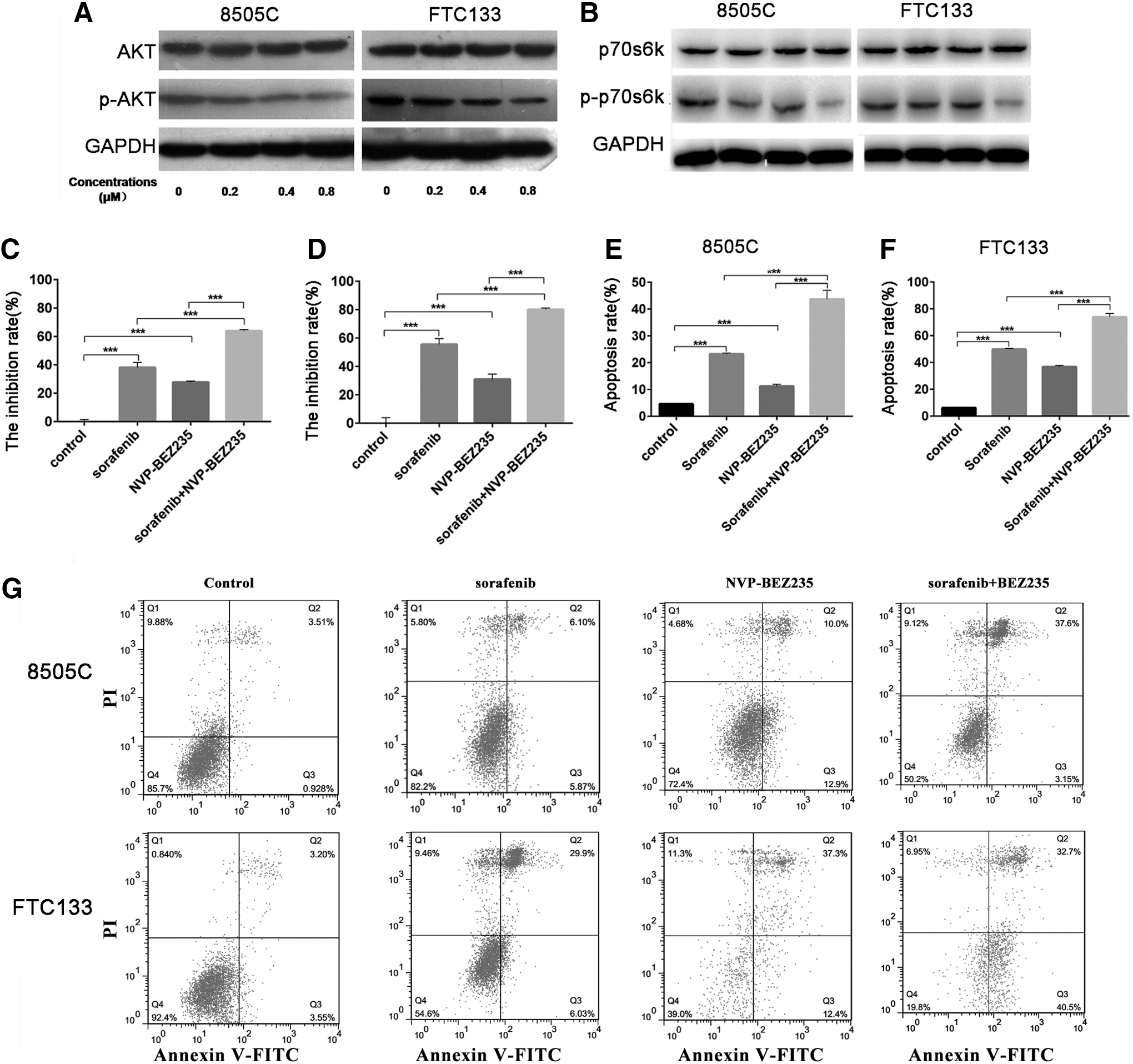
Combination treatment with sorafenib and BEZ235 significantly enhanced apoptosis and cell death.
As shown in Figure 4C and D, the viability of 8505C and FTC133 cells was suppressed by BEZ235. After incubation with 0.8 μM BEZ235 for 48 hours, the inhibition rates of 8505C and FTC133 cells were 38.1% and 55.4%, respectively (Fig. 4C, D). After incubation with both BEZ235 and sorafenib, the inhibition rates of 8505C and FTC133 cells were 63.9% and 80.1%, respectively (Fig. 4C, D). After the cells were incubated with BEZ235 for 48 hours, the rates of apoptosis of 8505C and FTC133 cells were 32.9% and 39.7%, respectively (Fig. 4E–G). After the cells were incubated with both BEZ235 and sorafenib, the rates of apoptosis of 8505C and FTC133 cells were 40.8% and 83.2%, respectively (Fig. 4E–G). The combined effects of BEZ235 and sorafenib improved the rates of apoptosis in both 8505C and FTC133 cells.
Discussion
Owing to the absence of effective therapeutic measures for radioactive iodine-refractory DTC, patients with this disease typically have a poor prognosis. 3,4 Sorafenib is a multiple kinase inhibitor that is used for clinical treatment in a variety of tumors; it is also associated with a good prognosis. 5,16 –20 A phase 3 clinical trial indicated that sorafenib led to a 5-month improvement in the median PFS compared with placebo in patients with progressive radioactive iodine-refractory DTC. 5 Although partial response (12.2% patients) and stable disease (74% patients, duration) as a result of sorafenib treatment still benefitted patients with radioactive iodine-refractory DTC, the median duration of treatment was only 10.6 months. 5 Primary and acquired resistance to sorafenib is still an obstacle in clinical practice, especially because little is known of the mechanism of sorafenib resistance in thyroid cancer.
Recent research has indicated that the development of thyroid cancer involves several signaling pathways and that the most important pathways in thyroid cancer development and in the proliferation of thyroid tumor cells are the MAPK (Ras/Raf/MEK) and PI3K/AKT/mTOR pathways. 9,10,21 An analysis of tumor samples from patients who participated in a sorafenib trial indicated that lower tumor expression of nuclear pAKT was associated with a partial response to sorafenib (p < 0.01). Several studies demonstrated that the MAPK pathway was inhibited by sorafenib and that another oncopathway, such as the PI3K/AKT/mTOR pathway, was also activated. 13,22,23
In this study, the cell viability assay indicated that sorafenib could inhibit proliferation in a different context in thyroid cancer cell lines. Sufficient evidence supports the idea that sorafenib inhibits the MAPK pathway through the suppression of proliferation and angiogenesis, as well as by the induction of apoptosis. 24,25 As the concentration of sorafenib increases, apoptosis levels also increase. The results of this study demonstrate that both ERK activity and AKT/mTOR activity were inhibited by sorafenib. As shown in the results, after incubation with sorafenib, the phosphorylation levels of both ERK and AKT were reduced. Phosphorylation of p70s6k, which is downstream effectors of AKT/mTOR, was also downregulated.
The role of AKT silencing in the treatment of thyroid cancer was demonstrated by RNA interference technology. After AKT was silenced by an RNA mimic, the apoptosis rate was markedly increased, and cell viability was inhibited in both the 8505C and FTC133 cell lines. Treatment with sorafenib combined with AKT silencing significantly enhanced the rate of apoptosis and reduced cell viability compared with a single treatment of sorafenib. Different studies have indicated that sorafenib suppresses the MAPK pathway and later stimulates or suppresses PI3K/AKT/mTOR pathway activity in different cell lines. 11 –14,22 In this study, sorafenib directly blocked the AKT/mTOR pathway in 8505C and FTC133 cells. These results at least partially explain why the AKT/mTOR pathway is inhibited by sorafenib and how other measures enhance the lethal effects of sorafenib.
The results of one clinical trial showed that sorafenib could effectively prolong PFS (10.8–21 months) and that sorafenib demonstrates promising clinical efficacy (partial response and stable disease) in cases of progressive metastatic or locally advanced radioactive iodine-refractory DTC. 26 –28 However, resistance to sorafenib was observed in most patients after 1 or 2 years of sorafenib administration. 26 –28 Therefore, alternative treatment measures should be considered for sorafenib-resistant patients who have previously undergone sorafenib treatment.
In vitro and in vivo studies of other cancer types have demonstrated that the addition of a PI3K/ATK pathway inhibitor to sorafenib elicits a better response than sorafenib alone.
13,29
–31
The combination of everolimus or temsirolimus and sorafenib in patients with advanced thyroid cancer who progressed on sorafenib alone is currently being assessed (
In this study, both MAPK and AKT/mTOR pathway activities were reduced by sorafenib. Sorafenib induced apoptosis and suppressed proliferation of thyroid cancer cell lines. The AKT/mTOR pathway was inhibited by RNA interference, and BEZ235 combined with sorafenib potently inhibited kinase targets, induced apoptosis, and inhibited proliferation. Therefore, AKT/mTOR inhibition might provide a complementary therapeutic benefit when combined with sorafenib. The combination of sorafenib and an AKT/mTOR kinase inhibitor could effectively enhance sorafenib treatment in vitro. However, the toxicity and clinical efficacy of this combination therapy remain to be evaluated.
Footnotes
Acknowledgments
This work was supported by the Zhejiang Province Natural Science Foundation of China (No. LY15H180002) and the National Natural Science Foundation of China (No. 81502318).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.