
Abstract
Aim:
The study investigated the ability of ataxia-telangiectasia mutated (ATM)/Rad3-related (ATR) signaling pathway to influence the proliferation, apoptosis, and radiosensitivity of nasopharyngeal carcinoma (NPC) cells.
Materials and Methods:
NPC tissues and corresponding adjacent normal tissues were collected from 143 NPC patients. The NPC CNE2 cells were assigned into a control group, X-ray group, CGK-733 group, and X-ray+CGK-733 group. The mRNA levels of ATM and ATR were evaluated using quantitative real-time polymerase chain reaction (qRT-PCR) and the protein levels of ATM and ATR using western blotting. The positive expression of ATM and ATR in tissues and nude mouse tumor tissues was determined by immunohistochemistry. Cell proliferation, migration, invasion, and apoptosis rates were analyzed by the 3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di- phenytetrazoliumromide (MTT) assay, scratch test, transwell assay, and flow cytometry, respectively. A nude mouse model of NPC was established to observe tumor volume and growth.
Results:
The mRNA levels of ATR and ATM and the expression of ATR and ATM protein in NPC tissues were significantly higher than those in adjacent normal tissues. The colony formation assay showed that the colony-forming rate decreased, showing radiation dose-dependent and CGK-733 concentration-dependent manners. Expression of ATM, ATR, Chk1, and Chk2 was evidently increased in the X-ray, CGK-733, and X-ray+CGK-733groups compared with the control group, and the aforementioned expression was highest in the X-ray+CGK-733 group among the four groups. The cell proliferation, invasion, and migration were decreased, tumor volume decreased and cell apoptosis increased in the X-ray, CGK-733, and X-ray+CGK-733 groups compared with the control group; the X-ray+CGK-733 group exhibited lowest cell proliferation, invasion and migration, smallest tumor volume, and highest cell apoptosis among the four groups.
Conclusions:
Inhibition of ATM/ATR signaling pathway reduces proliferation and enhances apoptosis and radiosensitivity of NPC cells.
Introduction
Nasopharyngeal carcinoma (NPC)
Ataxia-telangiectasia mutated (ATM) gene was primarily found in chromosome 11q22-23, and belongs to the phosphatidylinositol 3-kinase (PI3K) and PI3K-related protein kinase (PIKK) family, which also contains ATM and Rad3-related (ATR). 9,10 ATM and ATR signaling pathways are activated by DNA double-strand breaks and single-stranded DNA, respectively, and mediate cellular response to DNA damage. 11 In addition, ATM and ATR proteins are involved in human cancer, and inactivating mutations in ATM and ATR are characterized by susceptibility to cancer. 12 García et al. found that ATM/ATR-Chk1 pathway could induce cell apoptosis and cell cycle arrest and suppress tumor growth in prostate cancer. 13 Jung et al. also revealed that the ATM/ATR signaling pathway plays an important role in inhibiting tumor growth and cell cycle progression of colon cancer cells. 14 Moreover, it is reported that CX-5461 induces the arrest of G2-phase in cells, and p53-independent apoptosis was mediated by ATM and ATR signaling pathways. 15 A pervious study revealed that the inactivation of ATM/Chk2/p53 pathway contributes to the reduction of the radiation resistance in glioblastoma. 16 Therefore, the ATM/ATR signaling pathway may be a promising therapeutic target for NPC. Our study aims to explore the effects of the ATM/ATR signaling pathway on cell proliferation, apoptosis, and radiosensitivity of NPC cells.
Materials and Methods
Ethical statement
This study was approved by the clinical research Ethics Committee of the Tianjin Huanhu Hospital, and informed consents were obtained from all study subjects.
Subjects
The study included a total of 143 NPC patients, consisting of 93 males and 50 females (aged between 50 and 70 years), diagnosed in the Tianjin Huanhu Hospital from January 2012 to December 2014. According to the International Union against Cancer (UICC, 2008) tumor-node-metastasis (TNM) staging system, 17 patients were divided into the following three subgroups: 27 Stage I cases, 87 Stage II cases, and 29 Stage III cases.
Inclusion criteria of the study were as follows: patients with a complete medical history; patients diagnosed with NPC by specimen pathological examination; patients not receiving any chemotherapy, immunotherapy, surgery, or radiotherapy during or before the study; patients in good general condition attaining a Karnofsky Performance Scale (KPS) score of >70 points 18 ; patients having a white blood cell count over 4.0 × 109/L, platelet count over 100 × 109/L, and hemoglobin count over 120 g/L; and patients having normal hepatorenal functioning and normal electrocardiograms, and without severe adverse reaction to chemotherapy.
Exclusion criteria of the study were as follows: patients underwent chemotherapy in concert with radiosensitizer and radiation-protective agents; patients underwent surgical or chemoradiotherapy treatments; and patients with a history of other tumors.
Therapeutic regimens and efficacy evaluation
A 6 MV linear accelerator was used for fractionated radiotherapy five times per week for all patients, with an average dose of 74 Gy (70–78 Gy) for pars nasalis pharyngis, a dose of 66 Gy for cervical lymph node and a dose of 50 Gy for supraclavicular field. After treatment, the efficacy of radiotherapy was evaluated using magnetic resonance imaging of the nasopharynx based on the Evaluation Criteria in Solid Tumors (RECIST 1.1), 19 including complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). The sensitive group = CR+PR; the insensitive group = SD+PD.
Immunohistochemistry
NPC tissues and adjacent normal tissue samples were collected after biopsy. The obtained samples were fixed in 10% formalin and embedded in paraffin. The samples were sliced into slices of 3–4 μm thickness and attached to the glass slides coated with 3-aminopropyltriethoxysilane (APES) and baked in the oven at 60°C for 1–2 hours, and then placed at 4°C for future use.
After being dewaxed and hydrated conventionally, the tissue sections were placed in a 0.01 mol/L citrate buffer (pH 6.0) for antigen retrieval at high temperature and high pressure (pressure cooking 1–5 minutes, at 95°C with vacuum and negative pressure), and subsequently cooled at room temperature. The tissues samples were washed under running water and rinsed using phosphate buffered saline (PBS) three times (5 minutes each time), followed by the addition of 3% H2O2 into the solution. Subsequently, the tissue samples were cultured at room temperature for 10 minutes, and washed using PBS three times (5 minutes each time). Primary antibodies, including mouse anti-human ATM (diluted at a ratio of 1:75; Abcam, Cambridge, MA) and mouse anti-human ATR monoclonal antibody (diluted at a ratio of 1:75; Abcam) were added into the solution, followed by incubation in a refrigerator at 4°C for 8 hours. Next, the samples were washed using PBS three times (5 minutes each time), and supplemented with a second antibody (horseradish peroxidase (HPR)-labeled goat anti-rabbit IgG; ZSGB-Bio, Beijing, China) and cultured at room temperature for 15 minutes according to the instructions of the GTvision™ Kit.
Samples were washed again with PBS three times (5 minutes each time), and stained with diaminobenzidine (DAB; ZSGB-Bio) and counterstained with Hematoxylin (ZSGB-Bio), and the samples were sealed with neutral gum. Cell nuclei presenting light yellow, yellow, or brown particles were considered as the ATM/ATR-positive cells.
Results of stained sections were observed using a double-blind method by two pathologists uninformed about the clinical data. Five fields ( × 400) were randomly selected in the up, down, right, left, and middle areas of the uniformly stained section, and observed under a biological microscope (Olympus, Tokyo, Japan). The staining intensity of positive cells was observed under the five vision fields and the number of positive cells were counted and recorded. If the number of positive cells was ≥1000, the positive cell rate was calculated as the percentage of positive cells/total cells as follows: the positive cell rate <25% was considered as negative (−); the positive cell rate ≥25% was considered as positive (+).
Cell culture
The study included human NPC cell lines (CNE1, CNE2, HNE1, HONE1, and C666-1; Cancer Institute, Southern Medical University, Guangzhou, Guangdong, China), and immortalized nasopharyngeal epithelial cell lines (NP69; Cancer Institute, Southern Medical University, Guangzhou, Guangdong, China) were used as the control group. The cell lines were cultured in 1× Dulbecco's modified Eagle's Medium (DMEM) containing 10% PBS at 37°C with 5% CO2 and saturated humidity conditions. Cells were subcultured every 2–4 days. After two to three repetitions of subculture, the cells in the logarithmic phase of growth were used for further experiment. In addition, the mRNA expression of ATM and ATR in the six cell lines was detected by quantitative real-time polymerase chain reaction (qRT-PCR), and the NPC cell line with the highest ATM and ATR expression was selected for further experimentation.
Colony formation assay
The study used the following three doses of CGK-733 (an ATR/ATM signaling pathway inhibitor): 0, 5, and10 μm, 20 and the following four levels of radiation doses: 0, 2, 4, and 6 Gy. The CNE2 cells in logarithmic growth phase were washed using PBS two times, and digested to obtain a cell suspension. Once the cells were counted and diluted to appropriate concentration, the cells receiving different radiation doses were inoculated and cultured in a 60-mm diameter plate at 37°C with 5% CO2 under saturated humidity conditions for 2–3 weeks. The cultured cells were observed under a microscope every 2 hours, and the cell culture was terminated once a clone was visible to the naked eye. Once the supernatant was discarded, the cells were rinsed with PBS two times. Next, the cells were fixed with 5 mL pure methanol for 15 minutes, and consequently the fixative solution was discarded. Following the fixation, the cells were stained with Giemsa solution (Sigma-Aldrich Chemical Company, St Louis, MO) for 10–30 minutes, and washed under running water to remove the staining solution and dried in natural air conditions.
The plate was inversed and added with transparent film with mesh, the colony-forming units were counted under the naked eye, and the colony-forming rate was calculated and recorded. Colony-forming rate formula was as follows: (Colony-forming units/numbers of inoculated cells) × 100%. The experiment was conducted three times for each dosage.
Cell grouping
The CNE2 cells in logarithmic growth phase were assigned into the following four groups: the control group (without any treatment); the X-ray group (cells were exposed to a 6 MV X-ray radiation by a linear accelerator at a single dose of 6 Gy, radiation field of 15 × 15 cm, and source–skin distance of 100 cm, and cells were cultured for 24 hours); the CGK-733 group (cells were treated with 10 μm of CGK-733 [Abcam]); the X-ray+CGK-733 group (cells were exposed to 6 Gy X-ray radiation and treated with 10 μm of CGK-733).
Quantitative real-time polymerase chain reaction (qRT-PCR)
NPC tissues, adjacent normal tissues, and CNE2 cells in logarithmic growth phase were took, and then the total RNA were extracted using TRIzol reagent, and the RNA concentration was measured using the Eppendorf BioPhotometer (Eppendorf, Hamburg, Germany). Reverse transcription was performed using 3 μg RNA from each sample and cDNA synthesis was performed using the BcaBEST Kit (Invitrogen, Inc., Carlsbad, CA). The mRNA expression of ATR, ATM, checkpoint kinase 1 (Chk1), and checkpoint kinase 2 (Chk2) were detected using qRT-PCR techniques. Reaction conditions for qRT-PCR were as follows: predegeneration at 93°C for 2 minutes, then 40 cycles in total of 93°C for1 minute, 55°C for 1 minute, and 72°C for1 minute, and extension at 72°C for 7 minutes. Relative expression of genes was calculated using the 2−ΔΔCt method with glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) acting as the internal reference and based on the Ct value of target gene. Primers of ATR, ATM, Chk1, and Chk2 were designed based on the GenBank database using the Premier Primer 5.0 software. To avoid DNA contamination, upstream and downstream primers were designed as two exons. The GAPDH primer was designed by the ABI Research intelligence company (Oyster Bay, NY), and all primers were synthesized by the Shanghai Sangon Biotech Company (Shanghai, China) (Table 1).
ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia mutated and Rad3-related; F, forward; GAPDH, glyceraldehyde-3-phosphate-dehydrogenase; qRT-PCR, quantitative real-time polymerase chain reaction; R, reverse.
Western blot analyses
Once the cells were collected, obtained adherent cells were digested with trypsin. Next, the cells were centrifuged and washed twice, followed by cell lysis using a precooled cell lysing solution in ice for 30 minutes. Subsequently, the cell suspension was centrifuged at 13,000 rpm for 15 minutes at 4°C, and the supernatant were collected and transferred to an Eppendorf (EP) tube. With the addition of a 2 × sodium dodecyl sulfate (SDS) gel-loading buffer, the supernatant was boiled at 100°C for 5 minutes. The protein quantity of the sample was measured using the Bradford assay to ensure the uniformity of protein quantity, and then separated using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Next, the samples were transferred onto a polyvinylidene fluoride (PVDF) membrane, and supplemented with primary antibodies containing mouse anti-human ATM (diluted at a ratio of 1:3000, EP1890Y; Abcam) and mouse anti-human ATR monoclonal antibody (diluted at a ratio of 1:1000, 2B5; Abcam) for incubation at 4°C for 8 hours. The obtained samples were washed with PBS for 10 minutes × 3 times, and supplemented with second antibody HPR-labeled goat anti-rabbit IgG (at a ratio of 1:3000) and cells were developed using DAB. GAPDH was regarded as the internal reference, and the gel imaging system (Bio-Rad Laboratories, Hercules, CA) was used to detect the gray value of protein bands and analyze the relative protein expression of ATM and ATR.
3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di- phenytetrazoliumromide (MTT) assay
The CNE2 cells in logarithmic growth phase were washed with PBS three times, digested with trypsin, and resuspended using a RPMI-1640 medium (Roswell Park Memorial Institute medium; HyClone Thermo Fisher Scientific, Inc., Logan, UT). Next, the cells were counted and seeded into 96-well plates with a cell density of 5 × 103/well, and cultured in 100 μL of culture medium at 37°C with 5% CO2 in a constant temperature incubator. The medium was replaced after 6 hours and cells were cultured in an incubator for another 24 hours. After that, the medium was discarded, and the cells were washed with PBS twice and supplemented with a culture medium containing MTT (KeyGEN Biotech, Nangjing, Jiangsu, China). Once 50 μL MTT staining solution was added into each well, the cells was cultured in an incubator for 4 hours. Another 100 μL dimethyl sulfoxide (DMSO) was added into each well, and the plate was lightly shaken for 20 minutes. Subsequently, a microplate reader (DNM-9602G; Aolu Biotech, Shanghai, China) was used to measure the optical density value at a wavelength of 570 nm, and to calculate the proliferation rate of CNE2 cells.
Annexin-V fluorescein isothiocyanate/propidium iodide double staining
The CNE2 cells were cultured for 48 hours, and washed using PBS two times followed by digestion with trypsin without disodium edetate (EDTA) for 1 minute. The cells were collected after lightly blowing upon submerging in an RPMI-1640 medium containing 10% PBS. Next, the cells were centrifuged at a rate of 2000 rmp for 5 minutes and the supernatant was discarded, and subsequently the cells were washed and resuspended with 500 μL binding buffer. Cells were mixed with 5 μL Annexin V fluorescein isothiocyanate (FITC) (Biosea Biotechnology, Beijing, China) and 10 μL propidium iodide (PI), and cultured at room temperature without light for 5–15 minutes. Cell apoptosis in CNE2 cells was detected by flow cytometry (FACSAria III; Becton Dickinson, Oxford, United Kingdom) after 1 hour of cell culturing.
Scratch test
The CNE2 cells were inoculated into a 24-well plate after 24 hours. Upon reaching a cell confluence rate of 90%, a slide glass was scratched using a pipette tip (10 μL), and photographed using an inverted phase-contrast microscope (Axiovert 40C; Carl Zeiss, Oberkochen, Germany) at the central axis of each cell wall. The cells were cultured in an incubator at 37°C with 5% CO2 for 24 hours, and photographed again, and the scratch wound healing rate was measured. Triplicate wells were set up in each test.
Transwell assay
The CNE2 cell concentration was adjusted to 1 × 105/mL with a serum-free medium after transfection for 24 hours. A cell suspension (250 μL) was added into the upper chamber of transwell coated with Matrigel, and 800 μL medium with 15% fetal calf serum was added into the lower chamber of transwell for a 48-hour culture at 37°C. Cells in the lateral membrane of the upper chamber were removed and washed using PBS, and fixed in 4% formaldehyde for 10 minutes. The cells were observed and photographed under a microscope after Crystal Violet staining. Five visual fields were selected to count the number of apoptotic cells under a light microscope ( × 200), and the mean value of cell number was calculated and recorded. The experiment was repeated three times and invasion inhibition rate was calculated. The invasion inhibition rate (%) = (1−the number of invasive cells in the experimental group/the number of invasive cells in the control group) × 100%.
A nude mice model of NPC
Thirty-two BALB/c nude mice (16 males and 16 females) were purchased from the Institute of Laboratory Animal Science (Chinese Academy of Medical Sciences, Beijing, China). The mice were kept in a specific pathogen-free grade animal room with free access to water and food, and preadapted at natural light for 1 week. After 1 week, intragastric administration was conducted in experimental mice with an equal volume of normal saline, and was randomly stratified and identified using MICROSOFT EXCEL software. These mice were subcutaneously injected with CNE2 cells in the right axilla using 1 mL injections, and the cell suspension was prepared at a density of 1 × 106 cells in each 0.1 mL cell suspension. CNE2 cell suspension (0.5 mL) was injected into nude mice (∼5 × 106 cell per mouse) to establish NPC models.
After the NPC model establishment, all nude mice were divided into the control group (intraperitoneally injected with 10 μL of normal saline); the X-ray group (locally exposed to a linear accelerator at 6 Gy every 3 days, with a total dose of 24 Gy); the CGK-733 group (intraperitoneally injected with 10 μL CGK-733 every 2 days); and the X-ray+CGK-733 group (intraperitoneally injected with 10 μL CGK-733 every 2 days, and locally exposed to a linear accelerator at 6 Gy every 3 days after 24 hours, injected with CGK-733, with a total dose of 24 Gy). Seven days after tumor implantation, the long diameter (a) and short diameter (b) of tumor were measured at 12th, 14th, 16th, 18th, and 20th day by vernier caliper, and tumor size was recorded by the formula V = 1/2ab 2.
On the 20th day of the experiment, the infected nude mice were killed and tumor morphology was observed. The positive expression of ATM and ATR were evaluated by immunohistochemistry.
Statistical analyses
Statistical analyses were performed using the SPSS version 19.0 statistical software (SPSS Corp, Chicago, IL). Measurement data were expressed by mean ± standard deviation. Comparison of mRNA in NPC tissues and adjacent normal tissues was conducted by a t test. One-way analysis of variance was used for comparison among multiple groups, and least significant difference t test was used for comparison between the two groups. Enumeration data were expressed by percentage or ratio, and analysis by x2 test. p < 0.05 was considered to be statistically significant.
Results
The expression of ATM and ATR and their relationship with the radiosensitivity in NPC patients
Results of the qRT-PCR found that mRNA expression of ATM and ATR were significantly higher in the NPC tissues compared with the adjacent normal tissues (p < 0.05). Immunohistochemistry results show that positive expression of ATM presented as brownish in NPC tissues. Majority of the positive expression was found in the nucleus, some in the cytoplasm, and the staining intensity in the NPC tissues is stronger than the adjacent normal tissues. The positive rates of ATM and ATR (62.2% and 66.4%) in NPC tissues were significantly higher than positive rates (41.9% and 48.3%) in the adjacent normal tissues (both p < 0.05) (Fig. 1).
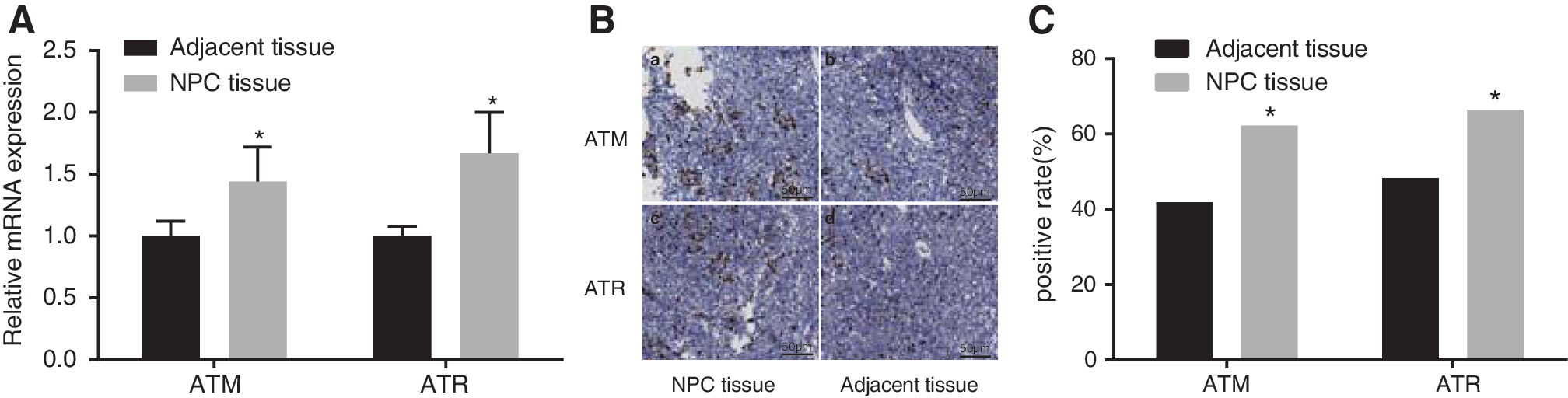
The expression of ATM and ATR between NPC tissues and adjacent normal tissues.
Among the 143 NPC patients, 108 cases (75.5%) were sensitive to radiotherapy, and 35 cases (24.5%) were insensitive to radiotherapy. Among the 89 patients with positive expression of ATM, 61 patients were sensitive to radiotherapy, and 28 patients were insensitive to radiotherapy. Among the 54 patients with negative expression of ATM, 47 patients were sensitive to radiotherapy, and 7 patients were insensitive to radiotherapy. Therefore, there were significant differences in the sensitivity to radiotherapy among the patients with positive and negative expression of ATM (p < 0.05). Among the 95 patients with positive expression of ATR, 65 patients were sensitive to radiotherapy, and 30 patients were insensitive to radiotherapy. Among the 48 patients with negative expression of ATR, 43 patients were sensitive to radiotherapy, and 5 patients were insensitive to radiotherapy. Similarly, there was a significant difference in the sensitivity to radiotherapy among the patients with positive and negative expression of ATR (p < 0.05). These results indicate that radiosensitivity in patients with positive expression of ATM/ATR was lower than patients with negative expression of ATM and ATR (Table 2).
ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia mutated and Rad3-related; CR, complete response; NPC, nasopharyngeal carcinoma; PD, progressive disease; PR, partial response; SD, stable disease.
The relationship between clinicopathological characteristics and the expression of ATM and ATR is shown in Table 3, and the results indicate that ATM expression was associated with lymph node metastasis and TNM staging and ATR expression was associated with lymph node metastasis, distant metastasis, and TNM staging (all p < 0.05); however, both ATM and ATM expressions were not associated with age and gender (p > 0.05).
ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia mutated and Rad3-related; TNM, tumor–node–metastasis.
CNE2 cell lines were selected for further cell treatment
mRNA levels of ATM and ATR were much higher in the NPC cell lines (CNE1, CNE2, HNE1, HONE1, and C666-1) compared with NP69 cell lines (p < 0.05) and consequently, the CNE2 cell lines were selected for further cell treatment (Fig. 2).

The mRNA levels of ATR and ATM in NPC cell lines (CNE1, CNE2, HNE1, HONE1, and C666-1) and nasopharyngeal epithelial cell line (NP69). *Compared with NP69, p < 0.05. ATM, ataxia-telangiectasia mutated; ATR, ataxia-telangiectasia mutated and Rad3-related; NPC, nasopharyngeal carcinoma.
A single X-ray dose of 6 Gy and a concentration of 10 μM CGK-733 were selected for further cell treatment
The results of colony-forming rate with different radiation doses are shown in Figure 3. The colony formation assay showed that the colony rate decreased, showing radiation dose-dependent and CGK-733 concentration-dependent manners. Thus, 6 Gy radiation dose and 10 μm CGK-733 were selected for further use. In addition, the results reveal that interactions between radiation and CGK-733 could inhibit cell colony formation (p < 0.05).
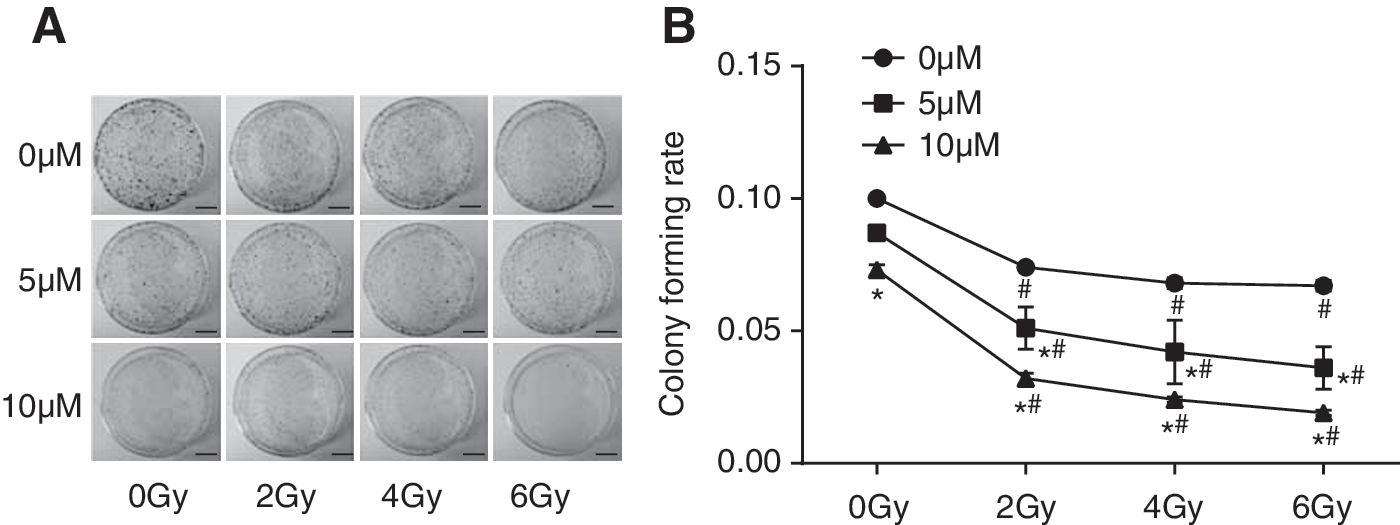
The colonies and colony-forming rates in CNE2 cells at 0, 5, and 10 μM CGK-733 after irradiation of 0, 2, 4, and 6 Gy.
The mRNA and protein levels of ATM, ATR, Chk1, and Chk2
Results of qRT-PCR and western blotting reveal that the X-ray, CGK-733, and X-ray+CGK-733 groups showed decreased mRNA and protein expression of ATM, ATR, Chk1, and Chk2 compared with the control group (all p < 0.05). The X-ray+CGK-733 group showed remarkably decreased mRNA and protein expression of ATM, ATR, Chk1, and Chk2 compared with the X-ray and CGK-733 groups (all p < 0.05). Moreover, there were no significant mRNA and protein expression differences between the X-ray and CGK-733 group (all p > 0.05) (Fig. 4).
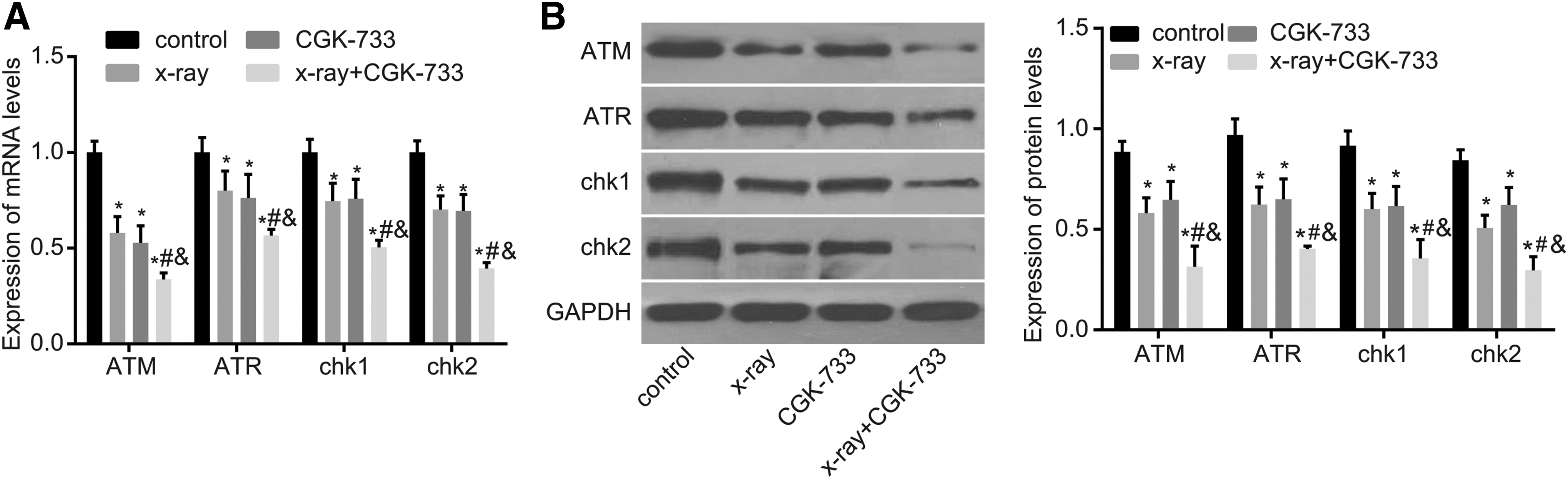
The mRNA and protein levels of ATM, ATR, Chk1, and Chk2 among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
X-ray irradiation with CGK-733 treatment reduced CNE2 cell proliferation
CNE2 cells in each group were cultured for successive 4 days, and the MTT assay was used to observe cell growth every day. The growth curve (Fig. 5) demonstrates that cell proliferation in the X-ray, CGK-733, and X-ray+CGK-733 groups significantly decreased compared with the control group after 48 hours (all p < 0.05). Compared with the X-ray and CGK-733 groups, cell growth velocity was the lowest in the X-ray+CGK-733 group (all p < 0.05).

Growth curve of CNE2 cells among the control, X-ray, CGK-733, and X-ray+CGK-733 groups. *Compared with the control group, p < 0.05; #compared with the X-ray group, p < 0.05; &compared with the CGK-733 group, p < 0.05. NPC, nasopharyngeal carcinoma.
X-ray irradiation with CGK-733 treatment facilitated CNE2 cell apoptosis
Cell apoptosis rates were detected by flow cytometry using Annexin-V FITC/PI double staining (Fig. 6), and the results show that the cell apoptosis rates in the X-ray group, CGK-733 group, X-ray+CGK-733 group, and control group were 17.3% ± 0.572%, 20.1% ± 0.617%, 31.8% ± 0.754%, and 10.1% ± 0.431%, respectively. Compared with the X-ray and CGK-733 groups, cell apoptosis rates most significantly increased in the X-ray+CGK-733 group (both p < 0.05). These results indicate that inhibition of ATM/ATR signaling pathway could promote cell apoptosis in CNE2 cells.
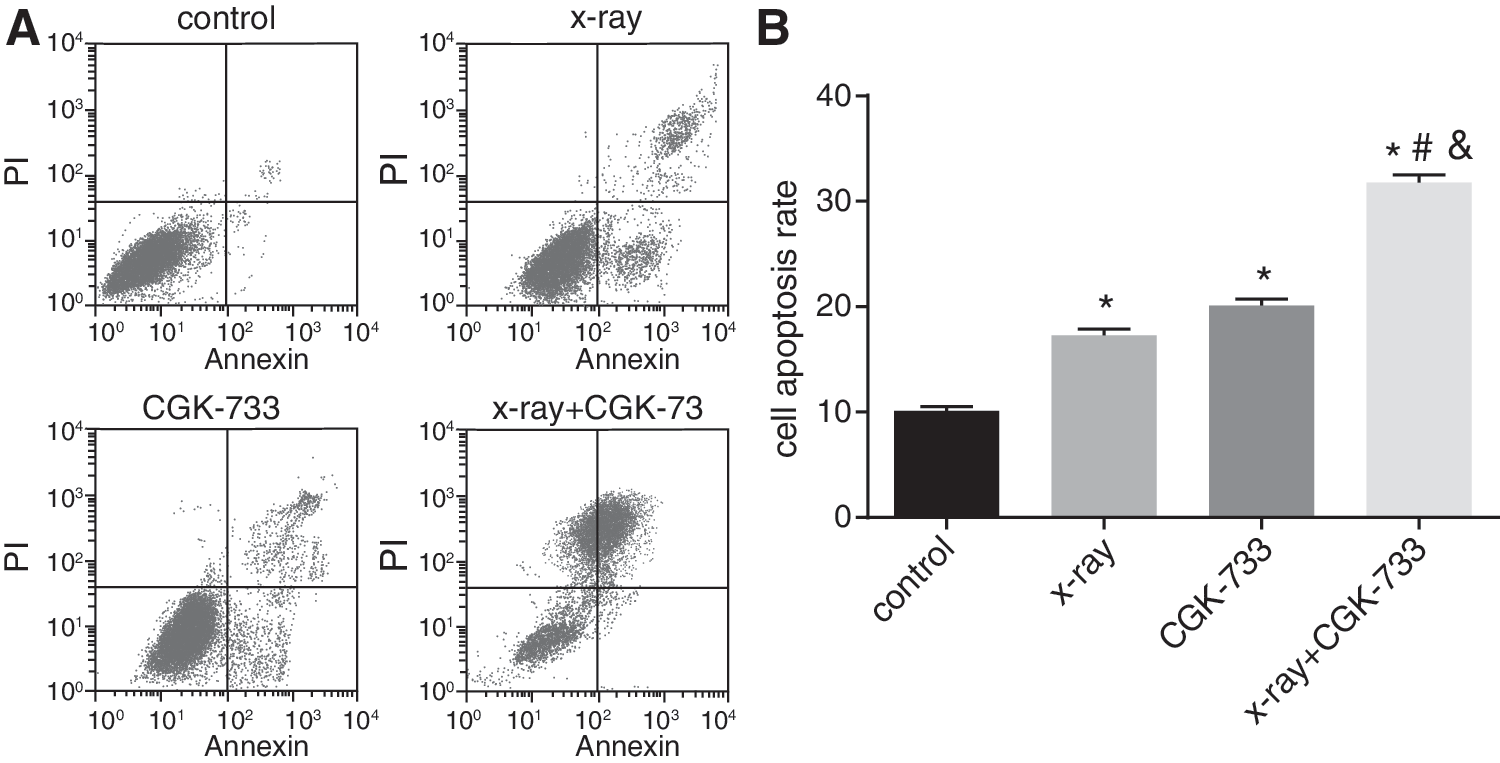
Cell apoptosis detected by flow cytometry among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
X-ray irradiation with CGK-733 treatment inhibited CNE2 cell migration
The results of scratch tests and cell migration ability are shown in Figure 7. After 24 hours of scratching, the X-ray, CGK-733, and X-ray+CGK-733 groups showed significantly increased scratch widths compared with the control group, and the best scratch widths were observed in the X-ray+CGK-733 group (all p < 0.05). In addition, the X-ray+CGK-733 group showed significant differences compared with the X-ray and CGK-733 groups (both p < 0.05).
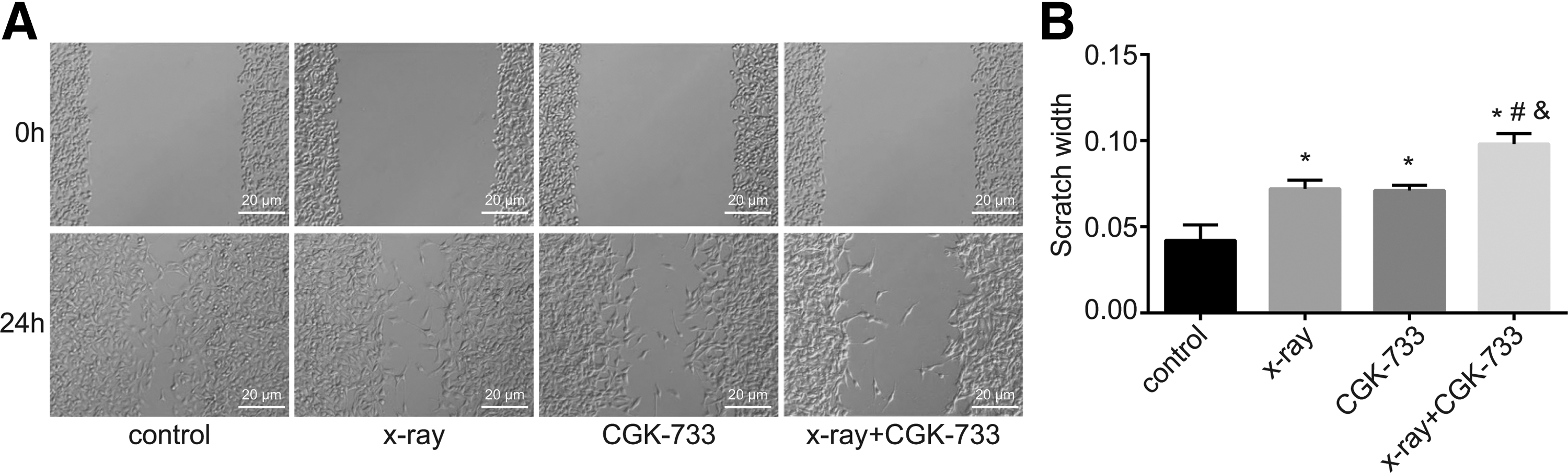
Cell migration detected by the scratch test among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
X-ray irradiation with CGK-733 treatment suppressed CNE2 cell invasion
The results show that the cell invasion ability in the X-ray, CGK-733, and X-ray+CGK-733 groups was much lower than the control group, with significant statistical differences (all p < 0.05). Furthermore, the cell invasion ability in the X-ray+CGK-733 group was significantly lower than the X-ray and CGK-733 groups (both p < 0.05) (Fig. 8).
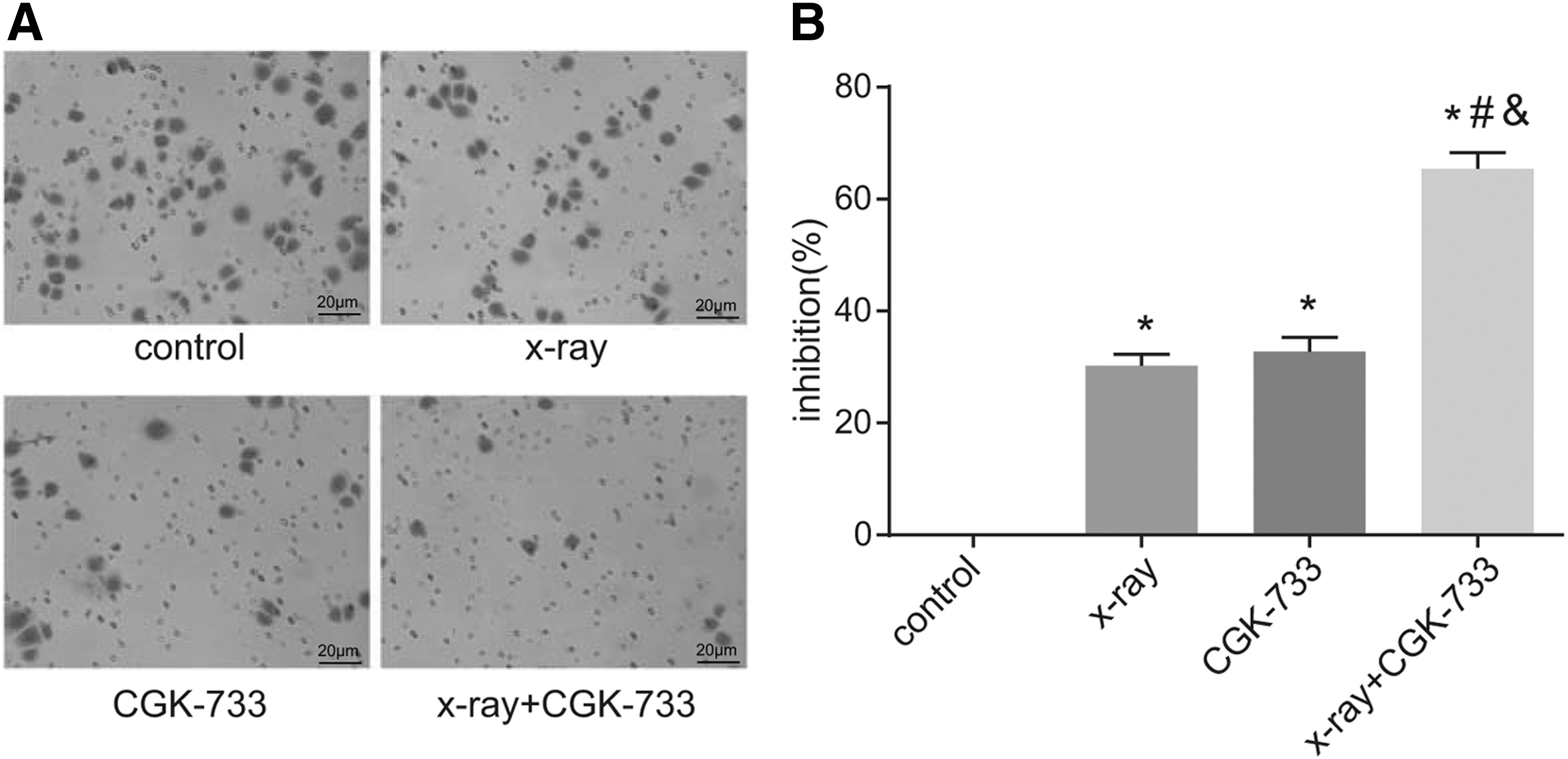
Cell invasion detected by the transwell assay among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
Tumor growth and volume in nude mouse after tumor implantation
After the NPC nude mice model was established, oval or irregularly shaped spherical tumors with thin pseudocapsule and fish meat-like surface were observed. At the 20th day of the experiment, the nude mice were killed and tumor formation in each group is shown in Figure 9. Tumor volume reduced and grew at a slower rate in the X-ray, CGK-733, and X-ray+CGK-733 groups compared with the control group (all p < 0.05). In addition, tumor volume in the X-ray+CGK-733 group was evidently decreased compared with the CGK-733 and X-ray groups (both p < 0.05).
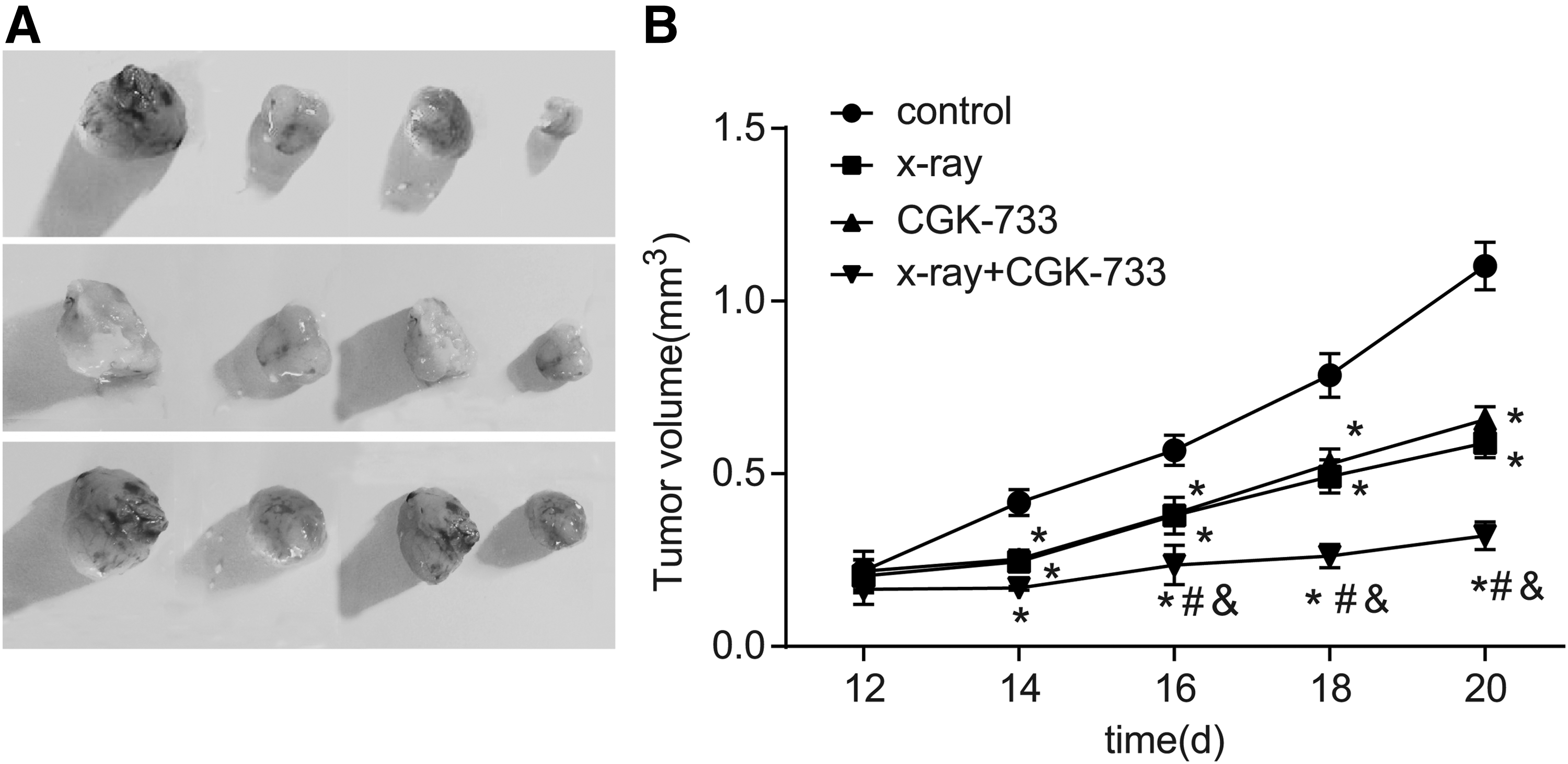
Tumor formation and growth curves of nude mouse among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
The expression of ATM and ATR in tumor tissues of nude mice
At the 20th day of the experiment, the nude mice were killed; results of immunohistochemistry show that positive expression of ATM and ATR were dark brown in color and primarily distributed in the cell nucleus (Fig. 10A). In the control, X-ray, CGK-733, and X-ray+CGK-733 groups, the positive rates of ATM were 87.5% (7/8), 50% (4/8), 62.5% (5/8), and 25% (2/8), respectively; whereas the positive rates of ATR were 100% (8/8), 50% (4/8), 50% (4/8), and 12.5% (1/8), respectively. The positive expression of ATM and ATR decreased in the X-ray+CGK-733 group compared with the control group (both p < 0.05), but there was no significant difference in the CGK-733 and X-ray groups (p > 0.05). Detailed data are shown in Figure 10B.
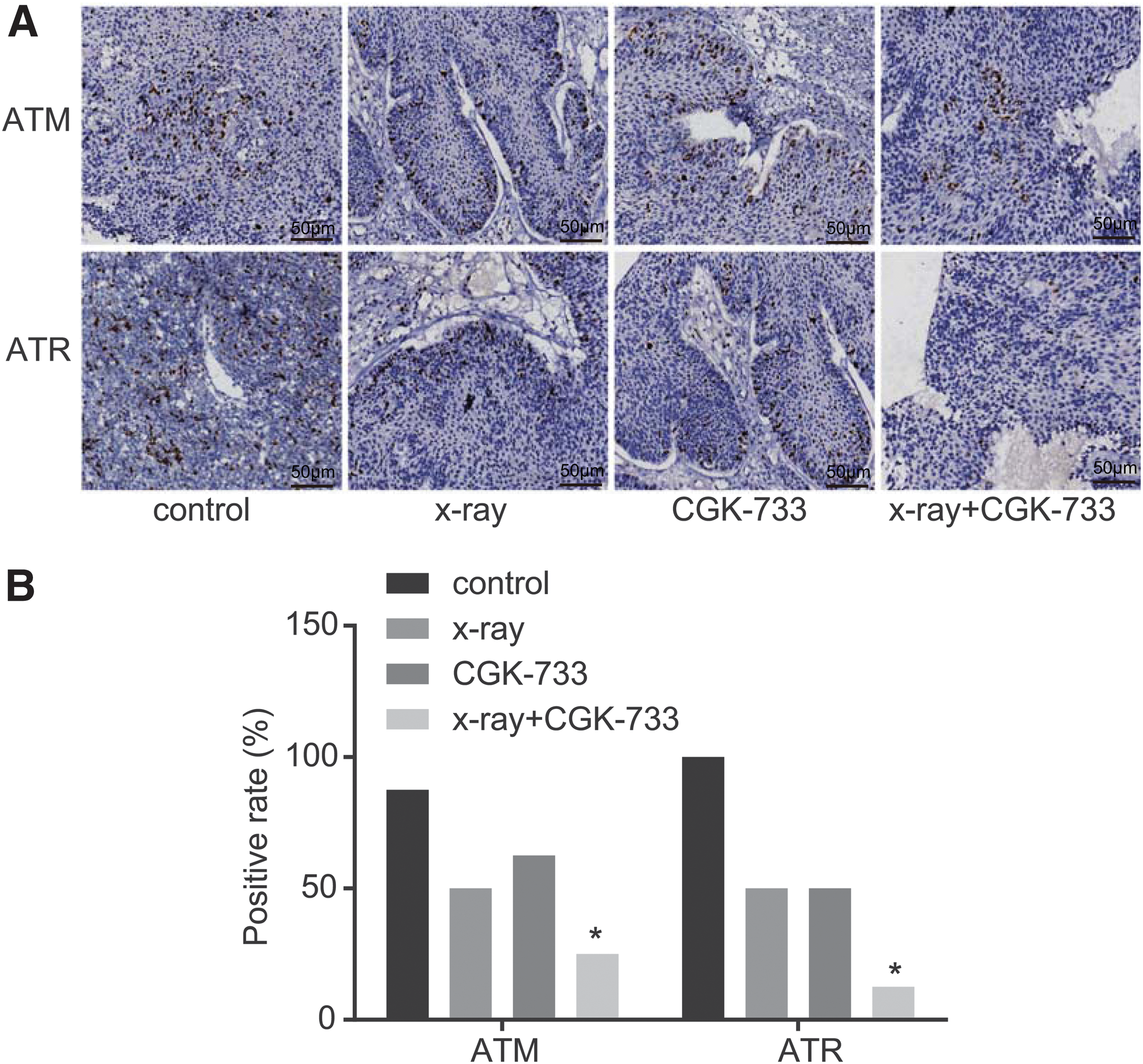
The expression of ATM and ATR detected by immunohistochemistry in tumor tissues of nude mice among the control, X-ray, CGK-733, and X-ray+CGK-733 groups.
Discussion
Recent research and studies have come to the realization that the ATM/ATR signaling pathway plays an important role in the prognosis and development of cancer. 21,22 Investigating the role of this signaling pathway in NPC may provide new biomarkers for the treatment of NPC. This is the first study aiming to investigate the role of ATM/ATR signaling pathway in the radiosensitivity of NPC cells, as well as NPC cell proliferation and apoptosis to our knowledge. Ultimately, our results show that the ATM/ATR signaling pathway could inhibit cell proliferation, promote apoptosis, and enhance radiosensitivity in NPC.
We observed a high expression of ATR and ATM in NPC cells and tissues. The ATM/ATR signaling pathway is commonly activated responding to different types of DNA damage. 13 Interestingly, ATM/ATR signaling pathway is overactivated in many human cancers, such as thyroid carcinoma and NPC. 23,24 This study also demonstrates that the lowest expression of ATM, ATR, Chk1, and Chk2 were found in the X-ray+CGK-733 group among the four groups. Activated ATR and ATM promote the phosphorylation of checkpoint kinases, including Chk1 and Chk2. 25 Both these kinases function similarly to mediate activities of the cell division cycle 25 homolog (CDC25) family. 26 Chk1 and Chk2 are phosphorylated by ATM/ATR in response to DNA damage or replication stress, resulting in the inactivation of CDC25s, thereby cell cycle arrested in lung cancer. 27 Activation of ATM/ATR-Chk2/Chk1 also promotes S and G2-M cell cycle arrest and inhibits cell apoptosis in cancer. 28 Studies have previously shown that high protein expression of ATM protein is correlated with worse prognosis of NPC. 24 The invasion, proliferation, and metastasis rate of NPC cell are quite high. Sun et al. reported that activation of ATM-mediated Snail promotes cancer cell invasion and metastasis in breast cancer. 29 Furthermore, the inhibition of ATM/ATR signaling pathway enhances apoptosis of prostate cancer cells by blocking DNA damage repair. 30 CGK-733, a specific ATM/ATR inhibitor, induces cancer cell death by blocking the kinase activities of ATM/ATR signaling. 31 In the present study, the ATM/ATR signaling pathway, inactivated as CGK-733, significantly inhibited the proliferation and invasion of NPC cells, and enhanced the apoptosis of NPC cells.
Additionally, results of the present study demonstrate that inhibition of ATM/ATR signaling pathway could enhance radiosensitivity in NPC patients. As a DNA damage sensor, nuclear ATM has been widely applied to mediate the sensitivity of tumor cells to DNA damage-inducing agents. Besides, the inhibition of ATM is also known as a target for cancer treatment with advantages over traditional PI3K/Akt inhibitors. 32,33 Moreover, ATM/ATR signaling pathway and Vγ2Vδ2 T cells are highly effective in treating ovarian cancer drug resistance. 34 ATM/ATR inhibitors are proven targets that cooperate with radiotherapy/chemotherapy, thereby enhancing cell apoptosis. 15 Lee et al. revealed that inhibition of ATM and ATR by CGK-733 could reduce cell adhesion regardless of cell exposure to ionizing radiation. 35 In this study, the inhibition of CGK-733 on ATM/ATR signaling pathway enhanced radiosensitivity in NPC. Principally, the suppression of ATM expression enhances radiosensitivity of radiosensitized NPC cells (CNE1) by cell cycle regulation. 36 Meanwhile, Li et al. found correlations between the inhibition of DNA double-strand break repair and radiosensitizing effect of NPC cells. 37 In the models of nude mice with NPC, tumor growth was inhibited by X-ray radiation treatment along with CGK-733, proving more effective than individual radiation treatment. In vitro and in vivo prove that the inhibition of ATM/ATR signaling pathway could enhance radiosensitivity of NPC cells.
Conclusions
In conclusion, the present study revealed that the inhibition of ATM/ATR signaling pathway could enhance apoptosis and radiosensitivity, and reduce proliferation of NPC cells in both in vivo and in vitro conditions. Our results provide evidence for the hypothesis that the inhibition of ATM/ATR signaling pathway combined with radiotherapy may be a promising target for the treatment against NPC. However, this study still has some limitations, for example, the best selection of ATM/ATR inhibitors is controversial, and also further study and analyses are required to explore the concrete mechanism of ATM/ATR signaling pathway in NPC.
Footnotes
Acknowledgment
The authors would like thank the reviewers for their helpful comments on this article.
Disclosure Statement
No competing financial interests exist.