
Abstract
Objective:
Gemcitabine, a nucleoside analogue, is used as a chemotherapeutic drug for the treatment of a wide variety of cancers. Therefore, radiolabeled gemcitabine may have potential as a radiotherapeutic agent for the treatment of various types of cancers. In the present work, an attempt has been made to radiolabel gemcitabine with 177Lu and study the preliminary biological behavior of 177Lu-labeled gemcitabine in tumor-bearing animal model.
Experimental:
Gemcitabine was coupled with p-NCS-benzyl-DOTA, a bifunctional chelating agent, to facilitate radiolabeling with 177Lu. The p-NCS-benzyl-DOTA-gemcitabine conjugate was radiolabeled with 177Lu, produced in-house and characterized by high-performance liquid chromatography. Tumor targeting potential of the radiolabeled agent was determined by biodistribution studies in Swiss mice bearing fibrosarcoma tumors.
Results:
177Lu-gemcitabine was prepared with a radiochemical purity of 95.7% ± 0.3% under the optimized reaction conditions. The radiolabeled agent showed adequate in vitro stability in normal saline as well as in human blood serum. Preliminary biological studies revealed rapid and significant accumulation of the radiotracer in the tumorous lesions along with fast clearance of activity from blood and other vital organs/tissue. Although tumor uptake gradually reduced with time, tumor to blood and tumor to muscle ratios were improved due to the comparatively faster clearance of activity from the nontarget organs/tissue.
Conclusion:
The present study demonstrates the preliminary potential of 177Lu-gemcitabine for targeted radiotherapy. However, further studies are warranted to assess its potential for radiotherapeutic applications.
Introduction
In recent years, linking of a therapeutic radionuclide to a chemotherapy drug, with an aim to either concentrate the chemocytotoxicity with the radiotoxicity into the tumor cell for enhancing efficacy or exploit the specific targeting properties to augment the therapeutic response, has emerged as an active and promising area of research in cancer therapy. This therapeutic paradigm, residing at the intersection of chemotherapy and radiation therapy, is expected to broaden targeted tumor therapy options in the foreseeable future. In view of this, development of radiotherapeutic agents for targeted tumor therapy, based on this concept, holds significant promise and deemed worthy of consideration.
While a myriad of factors contribute to the successful development of a targeted radiotherapeutic agent, selection of an appropriate chemotherapeutic drug with a broad spectrum of antitumor activity is a key determinant that decides its success. Gemcitabine (Gemzar®, 2′-deoxy-2′,2′-difluorocytidine), a synthetic pyrimidine nucleoside analogue and known antitumor substance, is commonly used as a chemotherapeutic drug and palliative agent for the treatment of a wide variety of cancers, such as nonsmall-cell lung cancer, pancreatic cancer, bladder cancer, ovarian cancer, soft tissue sarcoma, and metastatic breast cancer. 1 –10 It is also under investigation for the treatment of esophageal cancer and hematologic malignancies. 11,12
Gemcitabine is a potent inhibitor of deoxyribonucleic acid (DNA) synthesis and also inhibits various processes involved in the repair of genomic damages induced by radiation. 12 –16 Gemcitabine being an analogue of deoxycytidine gets activated inside the cell by deoxycytidine kinase to its active forms, gemcitabine diphosphate and gemcitabine triphosphate, which are actually responsible for its antineoplastic activity. 12,14 –17 It is reported that the diphosphate analogue of gemcitabine binds to the active sites of ribonucleotide reductase thereby irreversibly inactivating the enzyme. Once ribonucleotide reductase is inhibited, the cells stop producing the deoxyribonucleotides required for DNA replication and repair, eventually inhibiting the proliferation of cancer cells. 12,14 –19 Similarly, the triphosphate analogue of gemcitabine also gets incorporated into DNA and inhibits its replication, which in turn arrests the growth of tumor cells. 12,14 –16
Despite the fact that the clinical utility of gemcitabine as chemotherapy drug in the treatment of a variety of cancers either alone or in combination with other chemotherapy agent has been abundantly exploited, 1 –11 potential utility of radiolabeled gemcitabine in targeted tumor therapy is yet to be explored. Although radiolabeled gemcitabine is expected to have promising potential as an agent for targeted radiotherapy, realization of such an objective requires selection of an appropriate therapeutic radionuclide, conscious developments of defined chelate conjugation chemistry as well as radiolabeling strategy, in vitro stability assessment of the radiolabeled product, followed by biodistribution studies in animal model.
The usefulness of 177Lu as a radionuclide for targeted radiotherapy is already well established. 20,21 Lu-177 decays to stable 177Hf by emission of β− particles with a maximum energy of 497 keV, which is suitable for treating small- and medium-sized tumors. 20,22,23 It also emits gamma radiations (113 keV [6.4%] and 208 keV [11%]), which help in carrying out simultaneous pharmacokinetic and dosimetric evaluation. 24,25 The comparatively longer half-life of 6.73 days provides logistical advantages, which enables supply of 177Lu or 177Lu-based radiopharmaceuticals to geographically isolated centers with respect to the radioisotope production site or radiopharmaceutical formulation centers. 24 –28 Moreover, the possibility of producing this radionuclide in adequate quantity, sufficient specific activity, and high radionuclidic purity using medium flux research reactors makes 177Lu an attractive choice for developing agent for radiotherapeutic applications. 24 –28
Taking into account the wide-spectrum of antitumor activity of gemcitabine and advantages of using 177Lu as a radionuclide, an attempt was made to develop 177Lu-labeled gemcitabine for radiotherapeutic applications. Radiolabeling was effected through “indirect method” using a suitable bifunctional chelating agent (BFCA) owing to the difficulty encountered in direct incorporation of 177Lu in the gemcitabine moiety. Bifunctional chelator DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) or its suitable derivative is the obvious choice as BFCA for labeling with 177Lu owing to its ability to form thermodynamically stable complexes with Lu and these complexes exhibit high degree of kinetic inertness, which is one of the primary criteria for using the radiolabeled agents for clinical applications. 20,29 Therefore, for the present work, p-NCS-benzyl-DOTA [2,2′,2″-(10-(1-carboxy-4-((4-isothiocyanatobenzyl)-amino)-4-oxobutyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid] was chosen as the BFCA. The present article describes a systematic approach consisting of conjugation of p-NCS-benzyl-DOTA to gemcitabine moiety, optimization of radiolabeling conditions for formulation of 177Lu-labeled gemcitabine, evaluation of yield and radiochemical purity, assessment of in vitro stability and its preliminary biological evaluation in a tumor-bearing small animal model.
The primary objective of this study was the development of a radiolabeling strategy for the preparation of 177Lu-labeled gemcitabine and to assess its utility as a radiotherapeutic agent for targeted tumor therapy. With a view to accomplish the goal, a systematic approach was pursued and its outcome is documented in the present article.
Experimental
Materials and methods
Gemcitabine, used in the present study, was procured from Aldrich Chemical Company. p-NCS-benzyl-DOTA was obtained from macrocyclics. All the other chemicals used for the present study were procured from the reputed local manufacturers and were of “analytical reagent” (AR) grade. Proton nuclear magnetic resonance ( 1 H-NMR) spectra were acquired using a 300 MHz Varian VXR 300s spectrometer. Mass spectra were recorded on a QTOF Micromass instrument (Canada) using electron spray ionization (ESI).
Lu-177 (specific activity 15 ± 4.2 mCi/μg, 555 ± 155.4 MBq/μg), used for the present study, was produced in-house following the established procedure reported earlier. 26,28 All radioactive countings were performed using NaI(Tl) scintillation counter, obtained from Electronics Corporation of India Limited (India), by keeping the baseline and window at 150 and 100 keV, respectively, so as to utilize the 208 keV gamma photons of 177Lu, unless mentioned otherwise. The high-performance liquid chromatography (HPLC) analyses were performed using a dual pump HPLC system (JASCO 2080 plus, Japan), equipped with a radiometric detector system (Raytest, Germany) and C18 reversed phase HIQ-Sil column (5 μm, 4 × 250 mm). All the solvents used for HPLC studies were degassed and filtered before use and were of HPLC grade.
Biodistribution study was carried out in normal healthy Swiss mice, bred and reared in the laboratory animal facility of this institute, under standard management practice. The fibrosarcoma cell line, used for raising the tumors in the animals, was purchased from the National Center for Cell Sciences (India). Radioactive countings associated with the animal studies were carried out using a flat-type NaI(Tl) scintillation detector, procured from the Electronics Corporation of India Limited (India), by keeping the baseline and window at the same positions mentioned above. The animal study was approved by the institutional animal ethics committee and carried out in strict compliance with the relevant national laws related to the conduct of animal experimentations.
Statistical analysis of relevant data was performed by one-way analysis of variance using OriginPro® 8 software. Confidence level of 95% (p < 0.05) was taken for statistical significance.
Coupling of gemcitabine with p-NCS benzyl-DOTA
To couple gemcitabine with p-NCS-benzyl-DOTA, a mixture of p-NCS benzyl-DOTA (5 mg, 9.3 μmol) and gemcitabine hydrochloride (5 mg, 18.87 μmol) in 1 mL of double distilled water is magnetically stirred overnight at room temperature after adjusting the pH of the reaction mixture to 10 by slow addition of liquor ammonia. The progress of the reaction was monitored by thin-layer chromatography using 30% ethyl acetate in chloroform. After the completion of the reaction, solvent was evaporated under vacuum condition in a rotary evaporator. The solid product, thus obtained, was purified by preparative thin-layer chromatography and used for subsequent studies.
The purified conjugate was characterized by using standard spectroscopic techniques, such as 1 H-NMR spectroscopy and mass spectrometry.
1
H-NMR (DMSO-d6, δ ppm): 10.2 (4H, br s, -N-CH2-COO
ESI-MS (m/z): 814.81 (C33H44F2N8O12S, Calculated).
808.50 (M-6H) [Observed in negative scan mode].
Radiolabeling of gemcitabine with 177Lu
A stock solution of gemcitabine-p-NCS-benzyl-DOTA conjugate was prepared by dissolving 25 mg of the conjugate in 1 mL of ammonium acetate buffer (1 M, pH = 5). For radiolabeling, 200 μL of the stock solution (5 mg, 6.2 μmol) was added to 780 μL of ammonium acetate buffer and incubated with 20 μL of 177LuCl3 (1 mCi, 37 MBq) in a boiling water bath for a period of 1 hour.
Quality control of 177Lu-gemcitabine
HPLC studies were used for the characterization as well as determination of the complexation yield of 177Lu-labeled gemcitabine complex. HPLC analyses were carried out using water (A) and acetonitrile (B) mixed with 0.1% trifluoroacetic acid as the mobile phase using gradient elution technique (0–15 minutes 5% B to 75% B, 15–25 minutes 75% B, 25–30 minutes 75% B to 5% B, 30–35 minutes 5% B). About 20 μL of the radiolabeled preparation was injected into the column and elution was monitored by observing the radioactivity profile using an NaI(Tl) scintillation detector attached to the HPLC system. Flow rate was maintained at 1 mL/min.
Radiochemical studies
Various reaction parameters such as ligand concentration (gemcitabine-p-NCS-benzyl-DOTA conjugate), pH of the reaction mixture, and incubation temperature and period were varied over an appreciable range to arrive at the optimum protocol for the preparation of 177Lu-labeled gemcitabine-p-NCS-benzyl-DOTA conjugate with high radiochemical purity. The radiochemical purities of the 177Lu-labeled gemcitabine complex, prepared using different reaction conditions, were determined using the quality control technique mentioned above.
Preparation of inactive Lu-gemcitabine complex
To further characterize the lutetium-gemcitabine complex, an attempt was made to prepare the corresponding inactive complex using natural lutetium following the protocol mentioned below. A dilute solution of HCl (200 μL) containing Lu2O3 (0.5 mg) was added to gemcitabine-p-NCS-benzyl-DOTA conjugate (10 mg) dissolved in 800 μL of NH4OAc buffer (1 M, pH = 5) and the reaction mixture was incubated in a boiling water bath for 1 hour. After the completion of reaction, the solvent was evaporated and the product, thus obtained, was characterized by mass spectroscopy.
ESI-MS (m/z): 985.44 (C33H40F2N8O12SLu, Calculated).
986.28 (Observed) [in negative scan mode].
In vitro stability studies in normal saline and human blood serum
In vitro stability of 177Lu-labeled gemcitabine was studied by incubating the radiolabeled preparation (100 μL) separately with normal saline and normal human serum (400 μL). While the radiolabeled preparation was incubated in normal saline at room temperature till 6 days postpreparation, studies in human blood serum were continued for 2 days by incubating the mixture at 37°C. Radiochemical purity of the preparation was studied at various time intervals by aliquoting a small volume from the reaction mixture and subsequently carrying out the quality control analysis mentioned above.
In vitro serum binding studies
In vitro binding studies with human serum were carried out by incubating radiolabeled preparation (100 μL) with normal human serum (400 μL) at 37°C up to 2 days postpreparation. For determining serum binding, serum proteins were precipitated by adding equal volume of acetonitrile to it. The mixture was subsequently centrifuged at 8000 rpm for 4 minutes, and the radioactivity associated with the supernatant as well as the precipitate was counted separately in a well-type NaI(Tl) counter. Percentage serum binding was calculated from the counts observed in both the fractions.
Determination of partition coefficient (Log Po/w)
The partition coefficient (LogPo/w) of 177Lu-gemcitabine was determined in octanol/water system to evaluate the lipophilicity/hydrophilicity of the complex. For this, 200 μL of the radiolabeled preparation was added to a mixture of 800 μL of water and 1 mL of octanol and the resulting solution was vortexed thoroughly. This mixture was subsequently centrifuged at 3000 rpm for 5 minutes. Aliquots of 0.1 mL were withdrawn from both water and octanol layers and counted separately for 177Lu activity using the well-type NaI (Tl) counter. The Log Po/w values were determined from these data.
Biodistribution studies
Biological distribution and pharmacokinetic behavior of 177Lu-labeled gemcitabine were studied by carrying out biodistribution studies in Swiss mice bearing fibrosarcoma tumors. The tumors were developed by subcutaneous implantation of ∼1 × 106 cells in each animal, weighing 25–30 g. Tumors were allowed to grow until the diameter of the tumor reached ∼10 mm in size. Subsequently, the animals were intravenously injected with ∼100 μL of the radiolabeled preparation (∼100 μCi, 3.7 MBq) through one of the lateral tail veins. The animals were maintained in the normal laboratory environment with adequate normal diet and water till the designated time. The animals were sacrificed 3 hours, 1, 2, and 6 days postinjection using CO2 asphyxia. The organs were excised, washed with saline, dried, weighed in a weighing balance, and radioactivity associated with each organ/tissue was measured using a flat-type NaI(Tl) counter. Distribution of activity in different organs was calculated from these data and expressed as % injected activity (%IA) per unit mass of the organ/tissue. Three animals were used for each time point. The activity accumulated in the blood, skeleton, and muscle was calculated based on the assumption that these tissue/organs constitute 7%, 10%, and 40% of the total body weight of the animals, respectively. 30,31 Femur was chosen as the representative of bone for calculating the skeleton uptake. The excreted activity was determined by subtracting the activity accounted in all the organs from total IA.
Results
Coupling of gemcitabine with p-NCS benzyl-DOTA and its characterization
Gemcitabine-p-NCS-benzyl-DOTA conjugate was synthesized following a single-step reaction between gemcitabine and p-NCS-benzyl-DOTA in alkaline medium at room temperature. The scheme for the synthesis is shown Figure 1. The conjugate was characterized by 1 H-NMR spectroscopy. Further evidence in favor of its formation was also obtained from mass spectrometry.

Scheme for synthesis of gemcitabine-p-NCS-benzyl-DOTA conjugate.
Quality control study
Lu-177-labeled gemcitabine was characterized by HPLC using water and acetonitrile mixed with 0.1% trifluoroacetic acid as the mobile phase using gradient elution technique. The radiolabeled conjugate exhibited a retention time of 11.0 ± 1.0 minutes, while uncomplexed 177Lu eluted from the column within the void volume under identical conditions. A typical HPLC profile of 177Lu-labeled gemcitabine is shown in Figure 2.
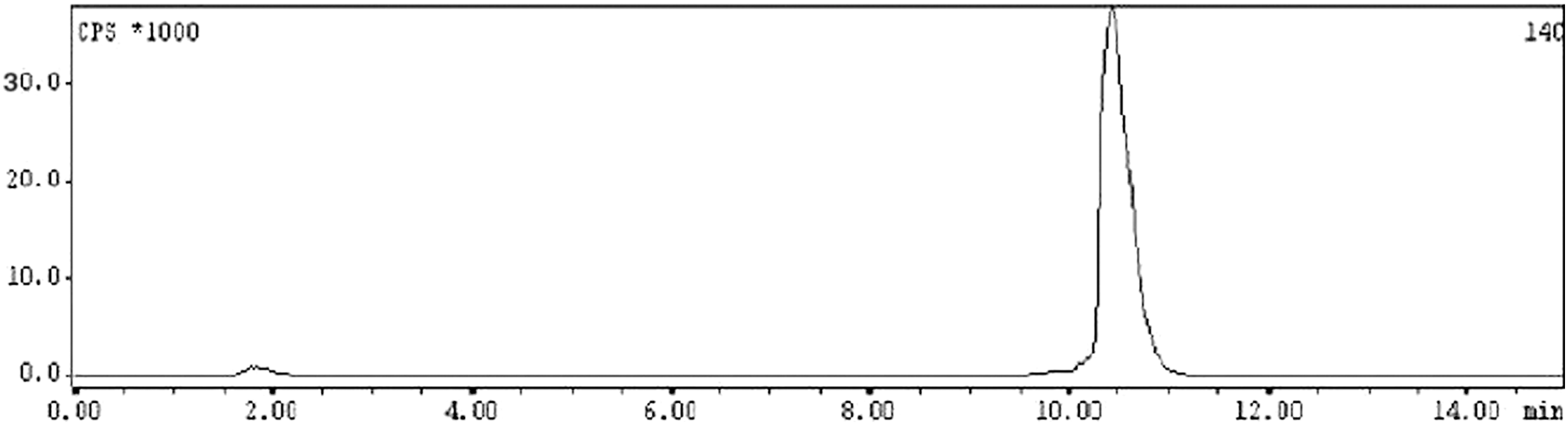
Typical high-performance liquid chromatography profile of 177Lu-gemcitabine.
Radiochemical studies
Different radiolabeling parameters such as concentration of gemcitabine-p-NCS-benzyl-DOTA conjugate, pH of the reaction mixture, incubation time and temperature were varied extensively to arrive at the optimum protocol for obtaining 177Lu-gemcitabine complex with high radiochemical purity. Ligand concentration, that is, concentration of gemcitabine-p-NCS-benzyl-DOTA conjugate was varied from 1 to 10 mg/mL and it was observed that while only 25.7% ± 1.2% radiolabeling yield was obtained when 1 mg/mL of ligand concentration was used, it increased to >95%, when the ligand concentration used was 5 mg/mL. Further increase in the ligand concentration did not yield any appreciable improvement in the radiolabeling yield of the complex, and hence, all the subsequent studies were carried out using a ligand concentration of 5 mg/mL. The variation of percentage radiolabeling yield with respect to the ligand concentration is shown in Figure 3.
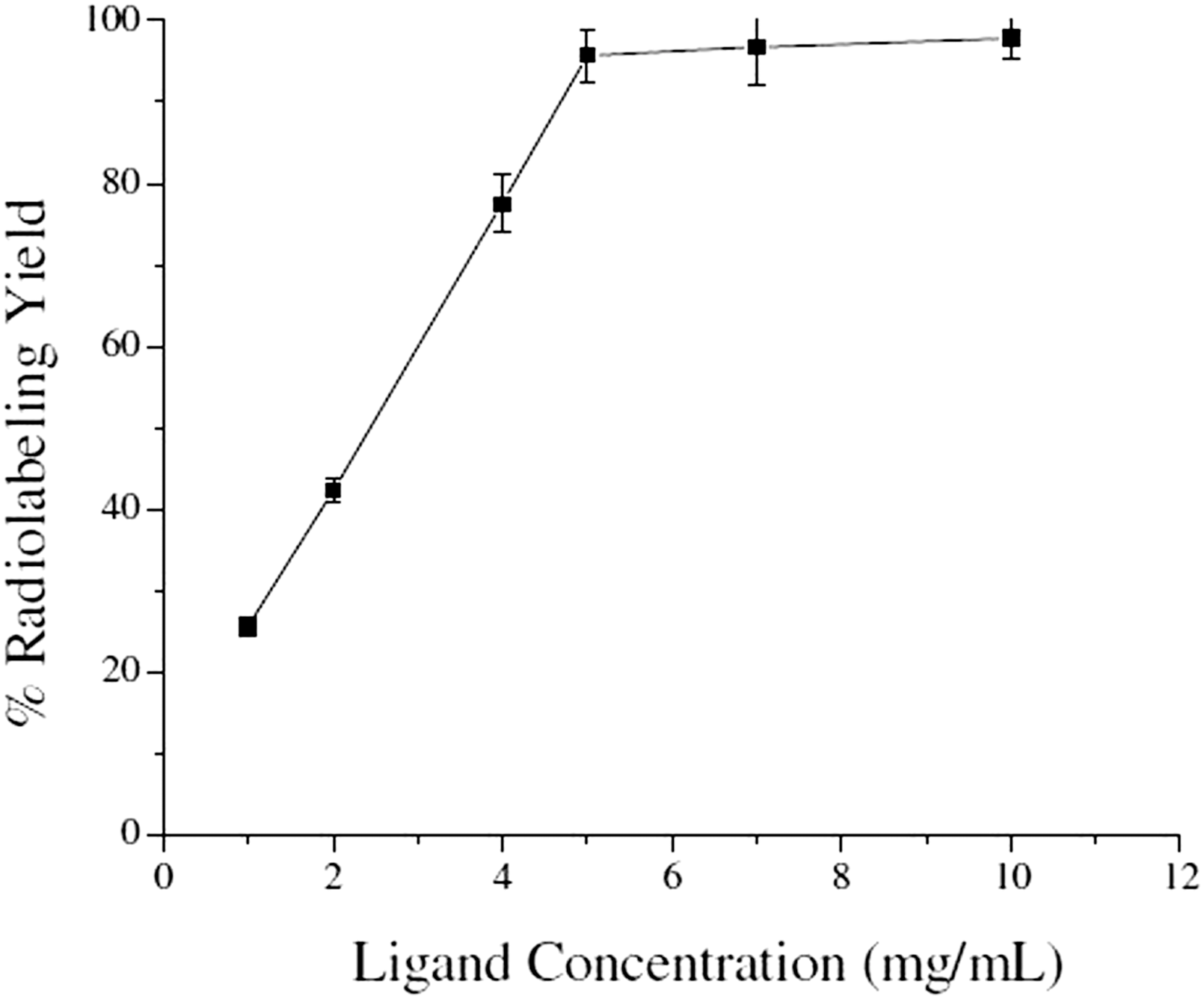
Variation of % radiolabeling yield of 177Lu-gemcitabine complex with ligand (gemcitabine-p-NCS-benzyl-DOTA conjugate) concentration.
To study the effect of pH on the complexation yield, pH of the reaction mixture was varied between 1 and 10, using either dilute HCl or NaOH solution and it was found that a maximum radiolabeling yield of 94.82% ± 1.89% could be obtained when the pH of the reaction mixture was maintained at pH ∼5. This is in agreement with the pH value reported in the literature for the preparation of various 177Lu-labeled agents, where the radiolabeling was effected through DOTA or its derivatives. 32,33 The radiolabeling was performed by incubating the reaction mixture in a boiling water bath, and the variation of percentage radiolabeling yield with respect to incubation period is shown in Figure 4. It is evident from this figure that percentage radiolabeling yield of 177Lu-gemcitabine becomes ≥95%, when the incubation is continued for 1 hour. Therefore, 1 hour incubation at 100°C was considered as the optimum incubation condition for the preparation of 177Lu-labeled gemcitabine.
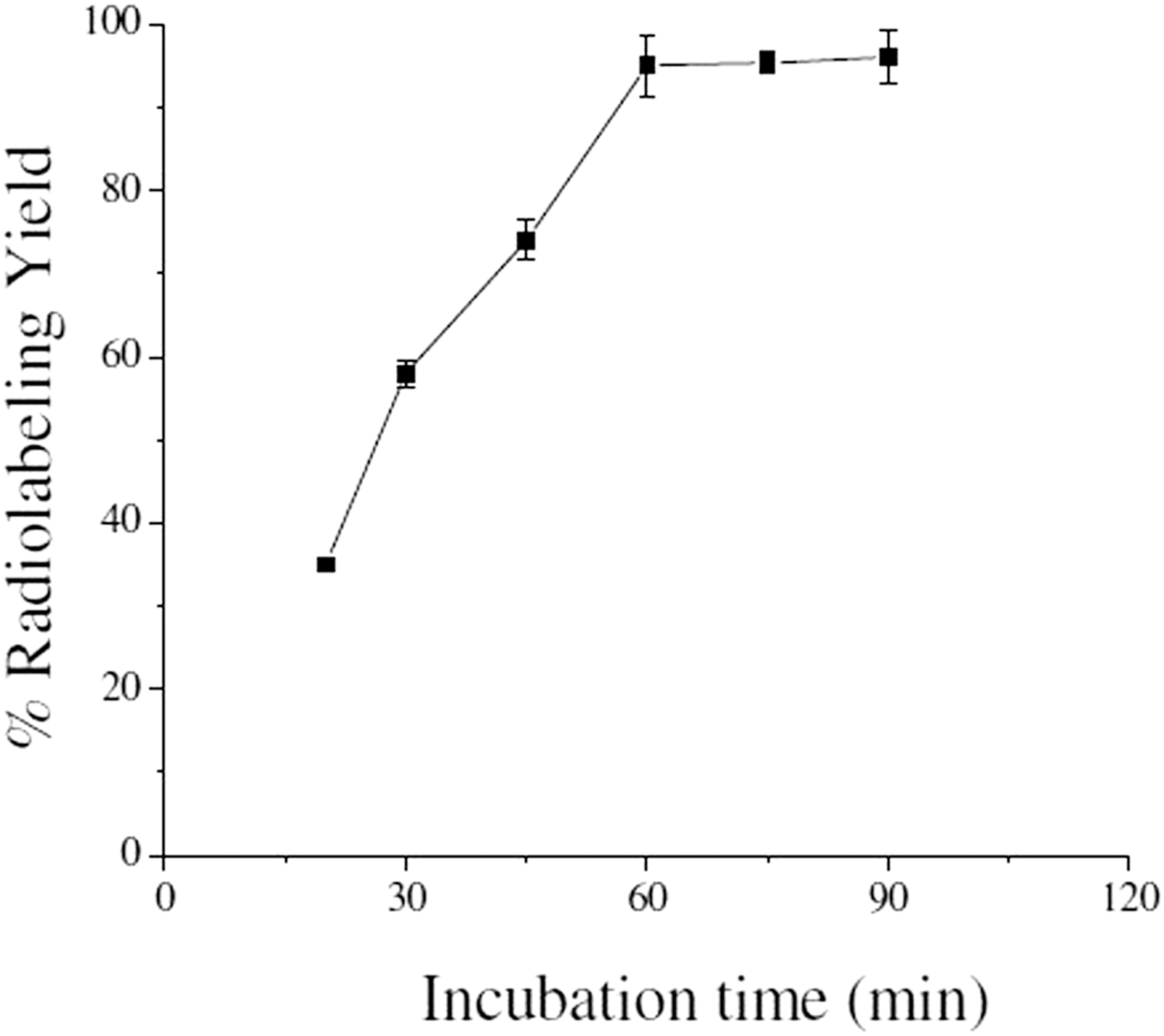
Variation of % radiolabeling yield of 177Lu-gemcitabine complex with incubation time.
Characterization of inactive Lu-gemcitabine complex
To further characterize the radiolabeled gemcitabine complex, corresponding inactive complex of Lu-gemcitabine was prepared and characterized by recording its mass spectrum. The observed m/z value of the Lu-gemcitabine complex (986.28) was found to be in close agreement with its theoretically calculated m/z value (985.44), which provided further evidence in favor of the formation of 177Lu-gemciatbine complex.
In vitro stability studies in normal saline and human blood serum
In vitro stability studies, carried out by incubating the 177Lu-gemcitabine in normal saline, showed that the radiolabeled conjugate retained its radiochemical purity of ≥90% till 6 days postpreparation, up to which the study was continued. On the contrary, 177Lu-gemcitabine maintained a radiochemical purity >65% till 2 days postpreparation when incubated with human blood serum at 37°C. The variation of radiochemical purity of 177Lu-gemcitabine complex with respect to the storage time in both normal saline and human blood serum is shown in Figure 5.

Stability of 177Lu-gemcitabine complex in normal saline at room temperature and human blood serum at 37°C.
In vitro serum binding studies
In vitro serum binding studies, carried out following the protocol mentioned above, indicated a percentage serum binding of 43.67 ± 5.35 for the 177Lu-gemcitabine complex, indicating moderate affinity of the radiolabeled preparation toward serum proteins.
Determination of partition coefficient (Log Po/w)
The partition coefficient (Log Po/w) of 177Lu-gemciatbine was determined following the protocol mentioned above and found to be −0.59. The value of Log Po/w indicates that the complex is hydrophilic in nature.
Biodistribution studies
The biological behavior of 177Lu-gemcitabine was studied by carrying out biodistribution studies using Swiss mice bearing fibrosarcoma tumors and results of the biodistribution studies are tabulated in Table 1. It is evident from the table that the agent showed considerable accumulation in the tumor (2.87% ± 0.72%) within 3 hours of administration along with fast blood clearance. No significant uptake of activity was observed in other vital organs/tissues at this time point, which is reflected in the comparative tumor to blood (99.48 ± 6.04) and tumor to muscle (26.15 ± 17.62) uptake ratios. The activity in the tumor was found to gradually decrease with time and 0.98% ± 0.03% of the IA was retained in per gram of tumor at 6 days postinjection. However, the uptake of activity, observed in different organs/tissues at the initial postadministration time point, was also diminished with time and this further improved tumor to blood and tumor to muscle ratios (452.06 ± 15.67 and 86.09 ± 8.93, respectively) at 6 days postadministration. The comparative uptake of the radiotracer in unit mass of different organs/tissues is shown in Figure 6, which clearly indicates the target specificity of the agent.
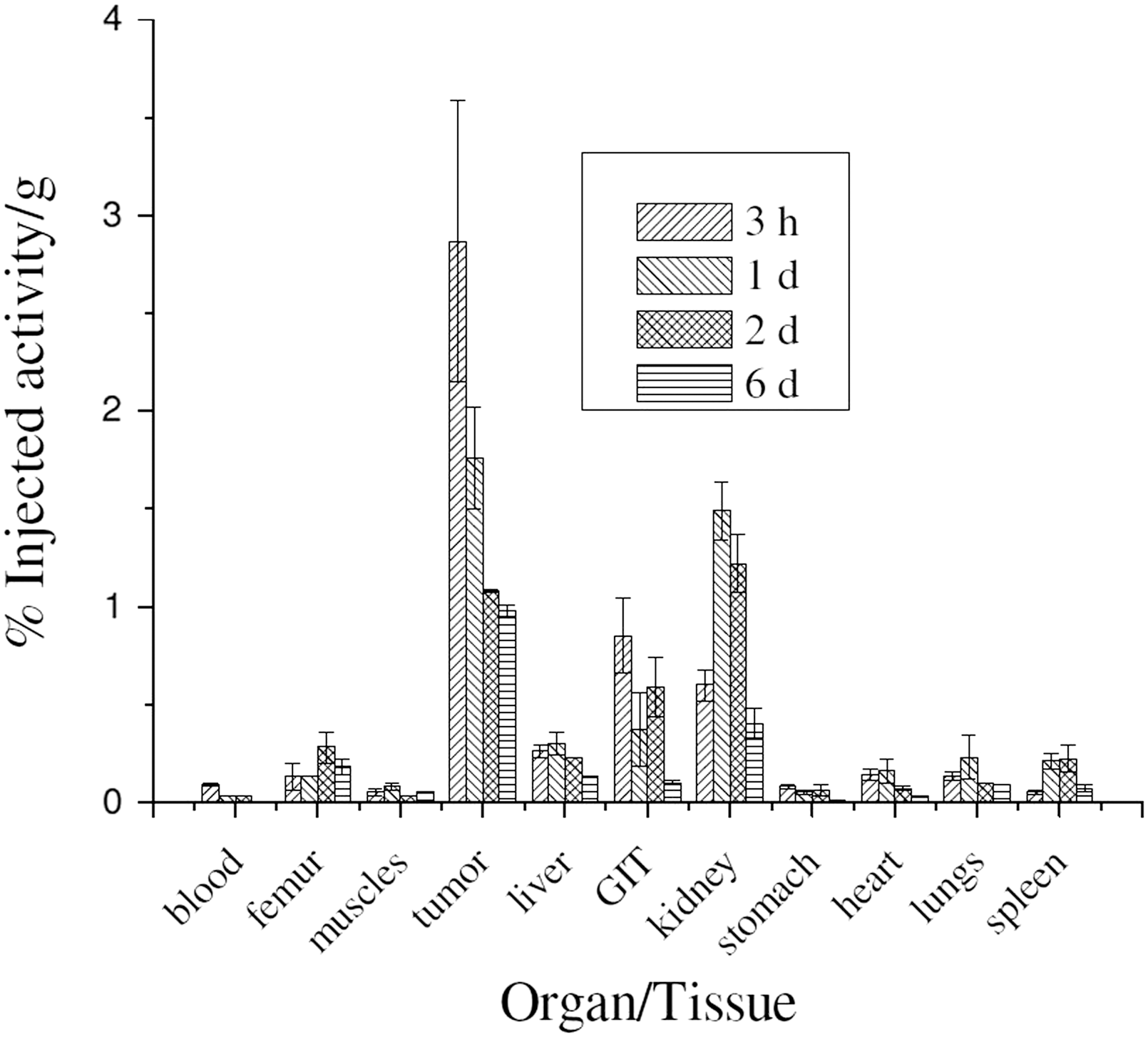
Comparative uptake of 177Lu-gemcitabine in unit mass of different organs/tissues of Swiss mice bearing fibrosarcoma tumors at 3 hours, 1, 2, and 6 days postadministration.
Discussion
Development of radiolabeled chemotherapeutic agents represents an expanding area in the quest for innovative approaches in tumor-targeted radionuclide therapy. 34 Although the clinical use of radiolabeled chemotherapeutic agent is still in its early stage, any breakthrough has the potential for far reaching clinical impact. With the appropriate selection of chemotherapy drug and therapeutic radionuclide pair, it may be possible to design radiotherapeutic agents adaptable for the treatment of a wide range of malignancies.
There appeared to enticing interest to consider the use of gemcitabine in cancer treatment owing to its commercial availability, documented antitumor activity as well as due to the fact that its use as an anticancer drug has already been approved by the U.S. Food and Drug Administration. 1,2,35 While antitumor properties of gemcitabine have been abundantly exploited in the treatment of a wide range of malignancies, 1 –11 its toxicity to normal tissues and acquisition of resistance by tumor tissues emerged as the major limitations that restrict its usefulness. 36 –39 One of the options to reduce drug dosing and toxicity to render gemcitabine more potent for cancer treatment would be its conversion into radiolabeled gemcitabine.
The authors' interest on the use of 177Lu as a therapeutic radionuclide was primarily due to their capability of in-house production of the radionuclide in required quality and quantity in “DHRUVA” reactor of their institute throughout the year in a cost-effective manner along with its relatively longer half-life, which minimizes the decay loss and its amenable chemistry, forming a metabolically resistant covalent bond with the BFCA of choice during the course of radiolabeling of gemcitabine. 20,21,24 –28 Moreover, the nuclear decay characteristics of 177Lu are quite suitable for developing agents for targeted radiotherapy. The presence of suitable energy gamma photons not only helps in simultaneous pharmacokinetic and dosimetric evaluation but also provides information regarding disease staging and finding out the efficacy of the radiotherapeutic modality. 25
One of the primary criteria that must be fulfilled for successful application of 177Lu-gemcitabine in targeted radiotherapy is that the binding between the 177Lu and gemcitabine should be essentially irreversible throughout the course of radiotherapy to prevent nontarget irradiation and its systemic effects. While the indirect method of radiolabeling using BFCA, which could coordinate with 177Lu to form stable complexes, is a successful and well-established model to preserve the biological activity of the targeting vector, selection of the BFCA and optimization of radioconjugation method are major determinants that dictate its success. Use of DOTA or its suitable derivative as BFCA for the formulation of 177Lu-based radiopharmaceuticals is a well-established and well-practiced procedure owing to the following considerations, namely (i) well-organized macrocyclic framework that facilitates formation of 177Lu complexes with extremely high thermodynamic stability and kinetic inertness, (ii) the low pKa values (2–5) of carboxylic groups that result in less competition from protons and minimum acid-assisted demetallation, and (iii) the high hydrophilicity of acetate chelating arms that assists faster clearance from blood, liver, and kidneys. 20,29,40,41 As the radiolabeling kinetics of DOTA-based 177Lu complex is normally very slow, the effect of experimental parameters in radiolabeling, including DOTA-gemcitabine conjugate concentration, pH, reaction temperature, and incubation time, was considered worthwhile investigating to arrive at the optimum conditions for the formulation of 177Lu-gemcitabine of desirable quality and yield. The optimized radiolabeling protocol arrived during the course of present work was prolific to prepare 177Lu-gemcitabine in good yields and acceptable purity. In vitro stability study was necessary to ensure the stability of the radiolabeled conjugate in the protein environment with which it interacts in blood and extravascular tissue spaces. The preliminary study showed that 177Lu-gemcitabine has adequate in vitro stability in human blood serum. Animal studies, carried out using fibrosarcoma tumor-bearing Swiss mice, showed rapid and encouraging accumulation of activity in the tumorous lesions along with fast clearance of the activity from the nontarget organs. The high target to nontarget ratios observed during the complete course of biodistribution studies indicated the preliminary potential of 177Lu-gemcitabine for targeted tumor therapy.
Conclusion
Gemcitabine, a chemotherapeutic nucleoside analogue, was coupled with p-NCS-benzyl-DOTA in a single-step process and the conjugate was radiolabeled with 177Lu with ≥95% radiochemical purity under the optimized reaction conditions. The radiolabeled preparation exhibited adequate stability in normal saline at room temperature and in human serum at 37°C. The radiolabeled complex showed significant tumor uptake (2.87 ± 0.72) %IA/g within 3 hours of administration and high tumor to background ratios throughout the time period of biological studies indicating its preliminary potential for targeted radiotherapy. However, further detailed investigations are warranted to determine the usefulness of the agent for radiotherapeutic applications.
Footnotes
Acknowledgments
The authors thank Dr. B.S. Tomar, Director, Radiochemistry and Isotope Group, Bhabha Atomic Research Centre (BARC), for his constant support and encouragement. The authors gratefully acknowledge the staff members of the Radiochemicals Section of Radiopharmaceuticals Division, BARC, for providing 177LuCl3 used in the present study. The authors thank Dr. Anupam Mathur, Board of Radiation and Isotope Technology, Navi Mumbai, for providing the facility of recording the mass spectra. The help rendered by the staff members of the Animal House Facility, Radiation Biology and Health Sciences Division, BARC, during animal experimentations is also acknowledged.
Disclosure Statement
The Bhabha Atomic Research Centre is a constituent unit of the Department of Atomic Energy, Government of India, and all research activities of the institute are fully funded by the Government of India. The authors declare that they have no conflict of interests and that there is no competing financial interest.